Alignment as a reflection of evolution. JalView
Last updated: 18-04-2017.
Files to download
[Download the whole .jvp project] [Download excel file used for Table 1]
[Download .fasta used in Task 2] [Download script used in Task 2]
[Download .fasta used in Task 3] [Download script used in Task 3]
Task 1. The main features of JalView
Main information about conservativity is assembled in Table 1 (the table is scrollable). It was received using JalView and Emboss program 'infoalign'. According to Table 1 it is possible to observe that sequence 'DNAK_PICTO/1-613' contains the biggest percent of absolutely conservative residues: 28,5 %. It also has the largest percent of absolutely functional-conservative residues: 45,7 %. In fact, this is an obvious observation, because the number of absolutely conservative residues and absolutely functional-conservative residues is the same for all sequences and 'DNAK_PICTO/1-613' is the smallest one. If we consider a more informative parameter 'amount and percent of 70% or more conservative residues', it can be seen that 'DNAK_MYCTU/1-625' sequence has the largest number of residues and their percentage: 347 and 55,5 % respectively. Several of absolutely functional-conservative residues are marked with a letter 'F' in Figure 1 (the figure is scrollable). In all three cases residues are interchangeable and able to perform the same functions (leucine, isoleucine and valine are aliphatic, nonpolar and uncharged (сolumns 53 and 54); lysine and arginine are both positively charged (column 87)). 'C' letter stands for 80% or more conservative residues, 'G' stands for positions containing gaps (Fig. 1).

| Sequence name | Domain | Sequence length | Alignment length | Amount of absolutely conservative residues | Percent of absolutely conservative residues | Amount of absolutely functional-conservative residues | Percent of absolutely functional-conservative residues | Amount of gaps | Percent of gaps | Amount of 70% or more conservative residues | Percent of 70% or more conservative residues |
|---|---|---|---|---|---|---|---|---|---|---|---|
| DNAK_PICTO/1-613 | Archaea | 613 | 730 | 175 | 24,0 % | 280 | 38,4 % | 16 | 2,2 % | 328 | 44,9 % |
| DNAK_METBF/1-620 | Archaea | 620 | 730 | 175 | 24,0 % | 280 | 38,4 % | 13 | 1,8 % | 343 | 47,0 % |
| DNAK_MYCTU/1-625 | Bacteria | 625 | 730 | 175 | 24,0 % | 280 | 38,4 % | 13 | 1,8 % | 347 | 47,5 % |
| DNAK_BIFLS/1-631 | Bacteria | 631 | 730 | 175 | 24,0 % | 280 | 38,4 % | 12 | 1,6 % | 342 | 46,8 % |
| GRP78_KLULA/1-679 | Eukaryota | 679 | 730 | 175 | 24,0 % | 280 | 38,4 % | 15 | 2,1 % | 318 | 43,7 % |
| HS71L_MOUSE/1-641 | Eukaryota | 641 | 730 | 175 | 24,0 % | 280 | 38,4 % | 14 | 1,9 % | 319 | 43,7 % |
Table 1. Parameters of conservativity.
Task 2. Unbelievable protein sequence evolution
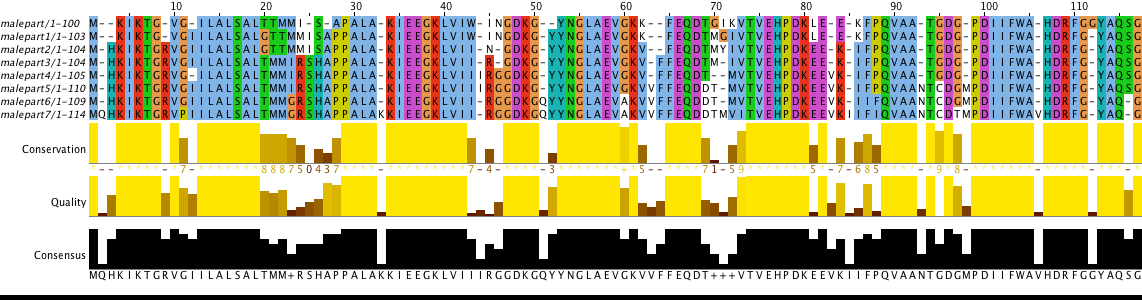
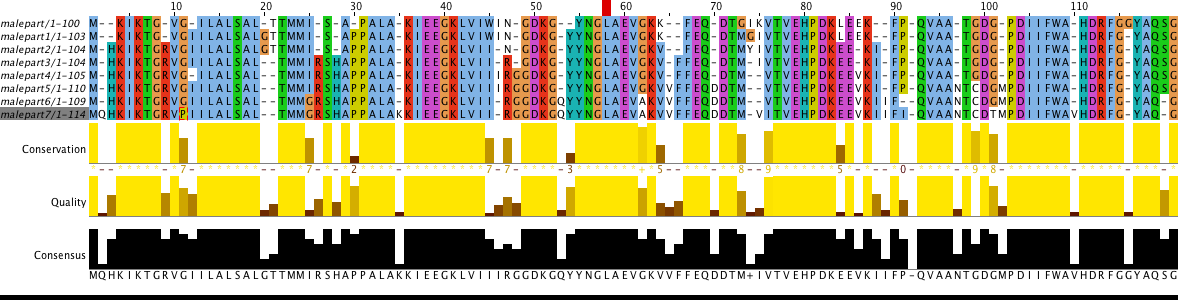
Part (1..100) of MalE (P19576) protein from Salmonella typhimurium (strain LT2 / SGSC1412 / ATCC 700720) was used for this task. Initial alignment of mutated protein is presented in Figure 2 and corrected alignment of mutated protein is presented in Figure 3. There is some mutations description (according to Fig. 3):
- position 2: glutamine insertion from p6 to p7;
- position 3: histidine insertion from p1 to p2;
- position 9: arginine insertion from p1 to p2;
- position 12: isoleucine insertion from p to p1, isoleucine deletion from p3 to p4, isoleucine insertion from p4 to p5;
- position 20: glycine insertion from p to p1, glycine deletion from p2 to p3;
- position 21: threonine deletion from p2 to p3;
- position 25: replacement of isoleucine with glycine from p5 to p6;
- position 26: arginine insertion from p2 to p3;
- position 28: histidine insertion from p2 to p3;
- position 30: proline insertion from p to p1;
Task 3. Unbelievable protein DNA sequence evolution
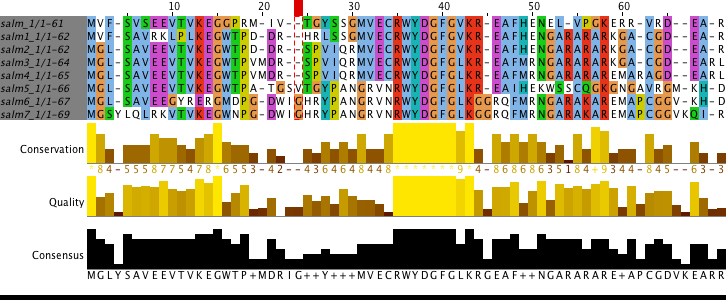
Protein CDS from Salmonella enterica subsp. enterica serovar Typhi str. P-stx-12 complete genome with coordinates CP003278.1:988749-988934 was used for this task. The results are shown in the Figure 4. As it can be seen it is almost impossible to correct this alignment due to the mutation was subjected to DNA and as a result there were shifts in the reading frame. In the course of the assignment, interesting difficulties arose: Emboss program msbar in the course of its work often created nonsense mutations which can't occur according to the task. That is why special script allowing to avoid nonsense mutations was created. Its basic logic is explained in Insertion 1.
msbar -count 7 -point 1 -block 0 salm(N).fasta salm(N+1).fasta -auto
descseq -sequence salm(N+1).fasta -outseq salm(N+1).fasta -name salm(N+1) -auto
transeq salm(N+1).fasta salm_dna(N+1).fasta -trim -auto
y=`grep -c "*" salm_dna(N+1).fasta`
while [ $y != 0 ]
do
msbar -count 7 -point 1 -block 0 salm(N).fasta salm(N+1).fasta -auto
descseq -sequence salm(N+1).fasta -outseq salm(N+1).fasta -name salm(N+1) -auto
transeq salm(N+1).fasta salm_dna(N+1).fasta -trim -auto
y=`grep -c "*" salm_dna(N+1).fasta`
doneInsertion 1. Script basic logic.
Task 4. Lecture statements discussion
Statement: mutations occur always and randomly
Actually some exceptions exist: prokaryotic SOS-reparation system in the case of serious damage works really fast but often makes mistakes. This is an example of choice between error tolerance and survival. Also, during the process of V(D)J rearrangement of lymphocyte DNA there is special enzyme 'terminal deoxynucleotidyl transferase' catalyzing the attachment of deoxyribonucleotides to the 3' end of DNA molecules.
Statement: mutations only in germ cells are inherited
Not necessary, mutations can occur, for example, in predecessors of germ cells. Also, not all mutations in germ cells can be inherited: for example, in male mitochondrial DNA.
Only genome nucleotide sequence evolves
Not entirely true statement: amino acid sequence evolves indirectly, too. Also, protein is the first step to check if mutations are positive, neutral or negative.
For proteins there is a check: the similarity of structures
That is not true for all proteins according to available research "Sequence-similar, structure-dissimilar protein pairs in the PDB[0].
Remark on the statements of the Great Craig Venter
“We built it from four bottles of chemicals.” Actually not. Of course it sounds beautiful, but another living cell was used to start the life: its lipid membranes, systems of transcription, replication and transport.
References
[0] Sequence-similar, structure-dissimilar protein pairs in the PDB, NCBI.