De novo genome assembly
Last updated: 10-12-2017.
Command table
| Command | Description |
|---|---|
| cat /P/y16/term3/block3/adapters/*.fa >> adapters.fasta | File containing adapters was created |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 SRR4240378.fastq 1.fastq ILLUMINACLIP:adapters.fasta:2:7:7 java -jar /usr/share/java/trimmomatic.jar SE -phred33 1.fastq 2.fastq TRAILING:20 MINLEN:30 | Deletion of adapters and reads trimming |
| velveth Assem 29 -short -fastq 2.fastq velveth Assem2 25 -short -fastq 2.fastq velveth Assem3 29 -short -fastq 2_7.fastq | k-mers preparation for full set of reads and half set |
| velvetg Assem -cov_cutoff auto velvetg Assem2 -cov_cutoff auto velvetg Assem3 -cov_cutoff auto | Assembly of full set of reads (k=29/k=25) and half set (k=29); cov_cutoff: removal of low coverage nodes |
| spades.py -s 2.fastq -o task6 --only-assembler -k 29 | Assembly using SPAdes |
| python /usr/lib/quast/quast.py task6/contigs.fasta | Program used for the analysis of results obtained from SPAdes |
Table 1. Used commands and their description.
Reads preparation
Step one (adapters removal): size before/after is 466360014/457540328 bytes; 81852 (1,85%) of reads were dropped, 4338735 (98,15%) survived.
Step two (reads trimming): size before/after is 457540328/438533540 bytes; 175703 (4,05%) of reads were dropped, 4163032 (95,95%) survived.
Assembly. Comparison of k=29 and k=25
Performed commands are presented in Table 1. According to Table 2, k=25 is a way more suitable for analysis due to max length of contigs and N50
| Parameter | k-mer length=29 | k-mer length=25 |
| N50 | 17169 | 27802 |
| Total length | 695 924 | 664 866 |
| Amount of contigs | 3 319 | 608 |
| The longest contigs | a)ID:8, lgth 68672, cov 27.7
b)ID:6, lgth 32730, cov 29.5 c)ID:19, lgth 29952, cov 30.7 | a)ID:6, lgth 70493, cov 44.7 b)ID:12, lgth 63787, cov 44.1 c)ID:9, lgth 56749, cov 45.2 |
Table 2. Assemblies comparison.
Analysis
Used commands are presented in Table 1.
ID:8, lgth 68672, cov 27.7
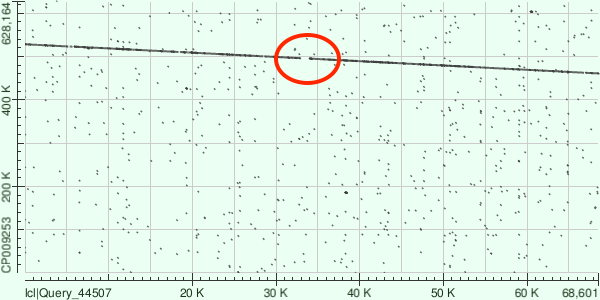
Contig's coordinates in genome are 460834-528275, amount of mismatches is 15253, gaps: 698. Dot matrix is presented in Fig.1. It's possible to observe small deletion in contig (marked red).
ID:6, lgth 32730, cov 29.5
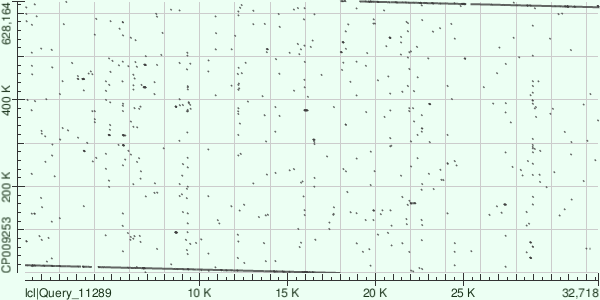
Contig's coordinates in genome are 15-17919 and 613659-627107 (beginning and end of genome), amount of mismatches is 6518, gaps: 221. Dot matrix is presented in Fig.2.
ID:19, lgth 29952, cov 30.7
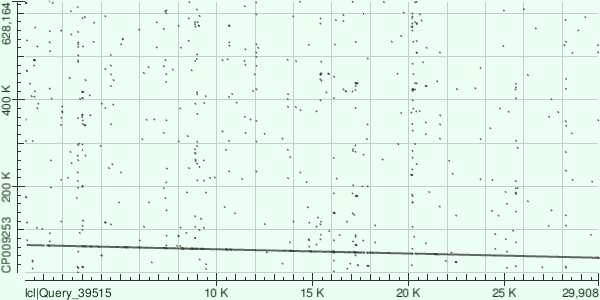
Contig's coordinates in genome are 35124-64577, amount of mismatches is 5851, gaps: 251. Dot matrix is presented in Fig.3. According to dot matrix and amount of mismatches and gaps, this contig seems like it has the best quality.
SPAdes
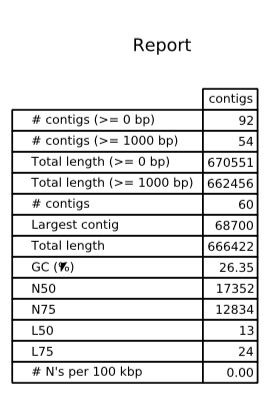
Used commands are presented in Table 1. Comparing to velveth, SPAdes is more user friendly and has more options. Also, it has nice pdf and html output, so I didn't have to waste time using excel (I used program called 'quast' (Fig.4)).
[Download full .pdf report].
Decreased amount of reads
For this task I have left only 2048673 of 4163032 reads. Results of assembly are not promising comparing to full reads set: N50 is 3953, maximum read length is 13789. But total length is not that bad: 643056.