EMBOSS
Last updated: 05-11-2017.
Task 1. Join fasta files
Initial data: [AAC74885.2.fasta] [AAC74886.1.fasta] [AAC74887.1.fasta] [AAC74888.1.fasta] [AAC74889.2.fasta] [AAC74890.1.fasta] [AAC74892.2.fasta] [AAC74892.1.fasta] [AAC74893.1.fasta]
Command:
seqret '*.fasta' pr9_1.fasta
Results: [pr9_1.fasta]
Task 2. Split fasta files
Initial data: [coding3.fasta]
Command:
seqretsplit coding3.fasta -auto
Results: [u00096.3_cds_aac77347.2_4310.fasta] [u00096.3_cds_aac77348.1_4311.fasta] [u00096.3_cds_aac77349.1_4312.fasta] [u00096.3_cds_aac77350.1_4313.fasta] [u00096.3_cds_aac77351.1_4314.fasta] [u00096.3_cds_aac77352.1_4315.fasta] [u00096.3_cds_aac77353.1_4316.fasta] [u00096.3_cds_aac77354.1_4317.fasta] [u00096.3_cds_aac77355.1_4318.fasta] [u00096.3_cds_aac77356.1_4319.fasta]
Task 3. Cut 3 CDS from .gb file
Initial data: [chromosome.gb] [coordinates]
Command:
seqret @coordinates pr9_3.fasta
Results: [pr9_3.fasta]
Task 4. Translate CDSs from fasta file
Initial data: [coding.fasta]
Command:
transeq coding.fasta pr9_4.fasta
Results: [pr9_4.fasta]
Task 5. Translate nucleotide sequence in six frames
Initial data: [coding.fasta]
Command:
transeq coding.fasta -frame 6 pr9_5.fasta
Results: [pr9_5.fasta]
Task 6. Change alignment format from .fasta to .msf
Initial data: [alignment.fasta]
Command:
seqret alignment.fasta msf::alignment.msf
Results: [alignment.msf]
Task 7. Print in output amount coincidental letters betwixt the second sequence of alignment and others
Initial data: [alignment.fasta]
Command:
infoalign alignment.fasta -refseq 2 -only -name -idcount -stdout -auto
Results:
PHZA_PSECL 90 VIBB_VIBCH 293 YRDC_BACSU 37 Y2499_AGRT5 44 Y2795_CAUCR 49 RUTB_ECO57 49 YECD_ECOLI 40 Y4030_CLOAB 34 YWOC_BACSU 46
Task 8. Сhange format from .gb to .gff
Initial data: [chromosome.gb]
Command:
featcopy chromosome.gb chromosome.gff
Results: [chromosome.gff]
Task 9. Extract from .gb file CDSs in fasta format and add description of protein function
Initial data: [chromosome.gb]
Command:
extractfeat chromosome.gb -type CDS -describe product pr9_9.fasta
Results: [pr9_9.fasta]
Task 10. Shuffle letters in given nucleotide sequence
Initial data: [coding.fasta]
Command:
shuffleseq coding.fasta pr9_10.fasta
Results: [pr9_10.fasta]
Task 11. Generate random sequence and find similar ones (E < 0.1) in GenBank
Notes: local nblast didn't work for me, so I used web version.
Initial data: [random1.fasta]
Command: (generation of random sequence)
makenucseq -amount 1 -length 1000 random1.fasta -auto
Results:
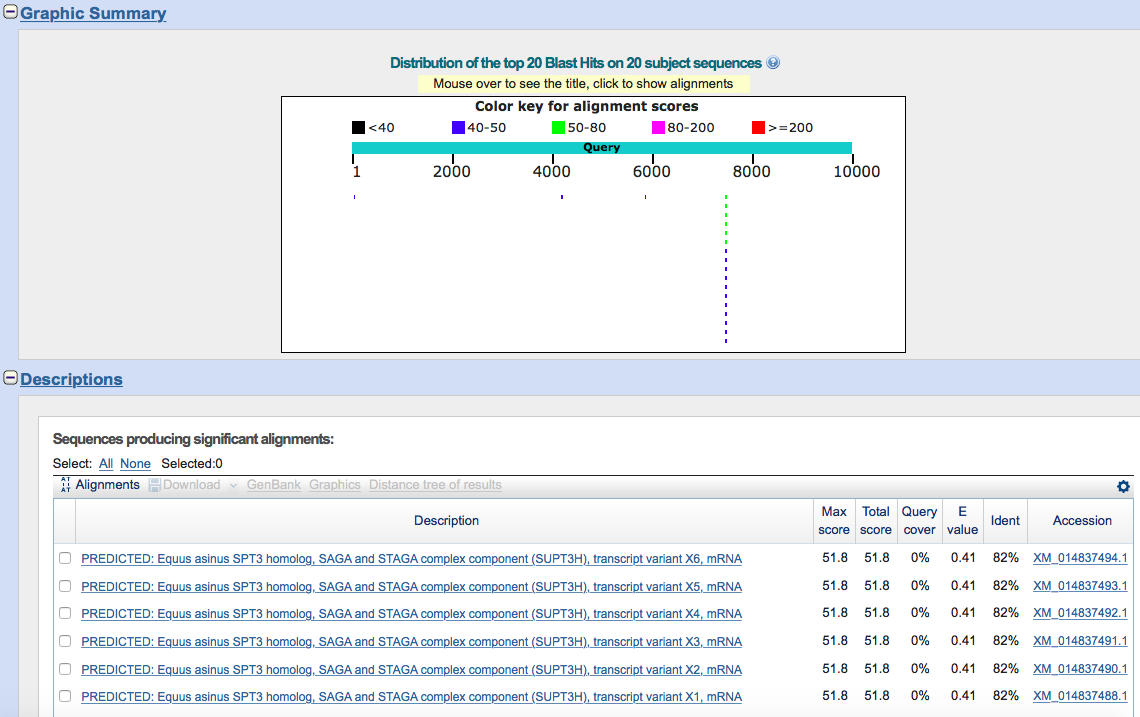
Task 12
Initial data:
Command:
Results:
Task 13. Frequencies of codons in given CDSs
Initial data: [coding.fasta]
Command:
cusp coding.fasta pr9_13.txt
Results: [pr9_13.txt]
Task 14. Frequencies of dinucleotides in the human chromosome, comparison of them with the expected
Initial data: [hs_ref_GRCh38.p7_chr22.fa]
Command:
wordcount hs_ref_GRCh38.p7_chr22.fa -wordsize 1 -outfile pr9_14_1.txt wordcount hs_ref_GRCh38.p7_chr22.fa -wordsize 2 -outfile pr9_14_2.txt
Results:
Results: [pr9_14_1.txt] [pr9_14_2.txt] [pr9_14.xlsx]
Task 15. Align CDSs according to their protein products alignment
Initial data: [gene_sequences.fasta] [protein_alignment.fasta]
Command:
tranalign gene_sequences.fasta protein_alignment.fasta -outseq pr9_15.fasta
Results: [pr9_15.fasta]
Task 16. Local multiple alignment of three nucleotide sequences
Initial data: [pr9_16_random.fasta]
Command:
makenucseq -amount 3 -length 5000 pr9_16_random.fasta -auto edialign pr9_16_random.fasta -outfile pr9_16.fasta -outseq pr9_16_seq.fasta
Results: [pr9_16.fasta] [pr9_16_seq.fasta]
Task 17. Delete gap symbols and other extraneous ones
Initial data: [alignment.fasta]
Command:
degapseq alignment.fasta pr9_17.fasta
Results: [pr9_17.fasta]
Task 18. Translation of end-of-line characters to unix format
Initial data: [pr9_13.txt]
Command:
noreturn pr9_13.txt pr9_18.txt
Results: [pr9_18.txt]
Task 19. Creation of three random sequences (length = 100)
Initial data: -
Command:
makenucseq -amount 3 -length 100 pr9_19.fasta -auto
Results: [pr9_19.fasta]
Task 20. Convert file from .fastaq format to .fasta
Initial data: [sra_data.fastq]
Command:
seqret sra_data.fastq fasta::sra_data.fasta
Results: [sra_data.fasta]