Поиск и описание полиморфизмов у пациента
Часть 1. Подготовка чтений.
Для подготовки чтений были взяты файлы из заданной директории. Далее была
произведена оценка качества чтений, для чего была выполнена команда:
fastqc chr21.fastq
Очистка чтений производилась с помощью программы Trimmomatic. Она была запущена
с такими параметрами, чтобы были удалены чтения с длиной менее 50 нуклеотидов и
с концов каждого прочтения были удалены нуклеотиды с качеством ниже 20.
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr21.fastq chr21_trimm.fast TRAILING:20 MINLEN:50
Число чтений до и после чистки
| Число чтений до чистки | Число чтений после чистки |
|---|
| 8158 | 7858 |
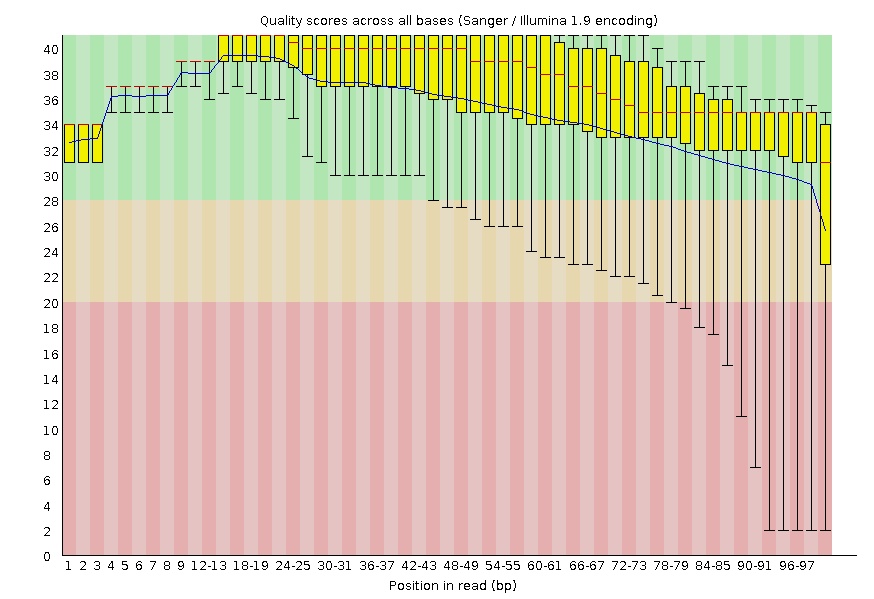
Картинки из FastQC "Per base quality"
До чистки
После чистки
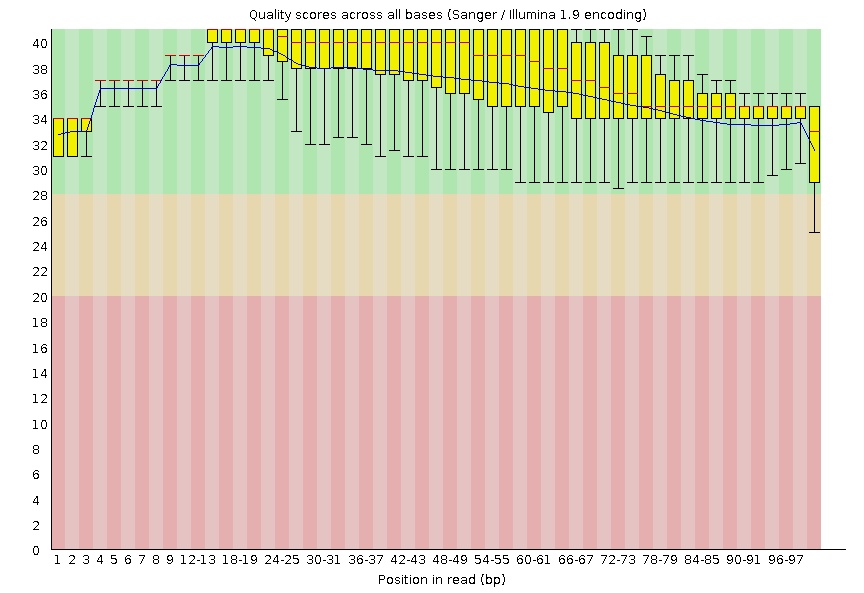 Стоит отметить, что число чтений после чистки уменьшилось, посколько
были удалены прочтения с длиной меньше 50. Также можем увидеть, что
улучшилось качество чтений(в желтой и красной области качества почти
ничего не осталось) поскольку с концов чтений были удалены
нуклеотиды с качеством ниже 20.
Стоит отметить, что число чтений после чистки уменьшилось, посколько
были удалены прочтения с длиной меньше 50. Также можем увидеть, что
улучшилось качество чтений(в желтой и красной области качества почти
ничего не осталось) поскольку с концов чтений были удалены
нуклеотиды с качеством ниже 20.
Часть 2. Картирование чтений
Картирование чтений было произведено программой hisat2, экспортированной с помощью команды
export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5
Таблица с командами и их описанием
| Команды | Описание |
|---|
hisat2-build chr21.fasta chr21_proc | Производит индексирование референсной последовательности |
hisat2 -x chr21_proc -U chr21_trimm.fastq --no-spliced-alignment --no-softclip>chr21.sam | Выравнивание прочтений и референса |
hisat2 -x chr21_proc -q chr21_trimm.fastq -S chr21.sam --no-spliced-alignment --no-softclip --met-file hisat2.txt | Сохранение выдачи в отдельный файл |
samtools view -b chr21.sam -o chr21.bam | Перевод в бинарный формат |
samtools sort chr21.bam chr21_sort | Сортировка выравниваний по координате в референсе начала чтения |
samtools index chr21_sort.bam | Индексирование отсортированного выравнивания |
samtools idxstats chr21_sort.bam>reads.txt | Запись числа откартированных чтений |
На геном откартировалось 7814 ридов, 46 ридов откартировано не было
Часть 3.1 Поиск SNP.
Создание файла с полиморфизмами в формате .bcf было выполнено с помощью команды:
samtools mpileup -uf chr21.fasta chr21_sort.bam > snp.bcf
Файл с отличиями в формате .vcf был создан с помощью команды:
bcftools call -cv snp.bcf -o snp.vcf
Было обнаружено 81 однонуклеотидный полиморфизм и 5 инделей.
Таблица с примерами полиморфизмов
| Координата | Тип | В референсе | В прочтении | Качество прочтения на участке | Глубина покрытия на участке |
|---|
| 16334658 | замена | C | T | 166.009 | 16 |
| 16334963 | замена | T | G | 225.009 | 75 |
| 43824106 | замена | A | G | 126.008 | 8 |
В целом можно сказать, что покрытие приведеных полиморфизмов сильно различается по качеству
Часть 3.2 Анализ SNP.
Аннотация SNP производилась с помощью программы ANNOVAR.
Индели были удалены перед запуском скрипта.
Подготовка входного файла была произведена с помощью команды:
perl /nfs/srv/databases/annovar/convert2annovar.pl.old - format vcf4 /nfs/srv/databases/ngs/simon_konnov/2/no_indel/snp.vcf > /nfs/srv/databases/ngs/simon_konnov/2/no_indel/snp.avinput
Refgene
Аннотация по генам была произведена с помощью команды:
perl /nfs/srv/databases/annovar/annotate_variation.pl.old -out rs.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb.old/
В результате быле получены файлы с описанием всех полиморфизмов, описанием синонимичности и log-файл.
База данных Refseq делит snp по их локализации.
Количество snp: exonic - 4,intronic - 68, UTR3 - 9.
| Ген | Число SNP |
|---|
| NRIP1 | 15 |
| UBASH3A | 25 |
| AGPAT3 | 41 |
rs есть у 60 snp.
Dbsnp
Использовалась команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl.old -filter -out rs.snp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb.old/
Аннотация показала, что 62 полиморфизма имеют rs, а 25 не имеют
1000 genomes
Использовалась команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl.old -filter -out rs.1000genomes -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb.old/
Было отобрано 58 полиморфизмов, а 29 попали в filtered
Gwas
Использовалась команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl.old -filter -out rs.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb.old/
Найденные snp не связаны с фенотипическими признаками
Clinvar
Использовалась команда:
perl /nfs/srv/databases/annovar/annotate_variation.pl.old -filter -out rs.clinvar -dbtype clinvar_20150629 -buildver hg19 snp.avinput /nfs/srv/databases/annovar/humandb.old/
В выдаче не было ни одного snp, связанного со здоровьем человека.
© Simon Konnov 2017