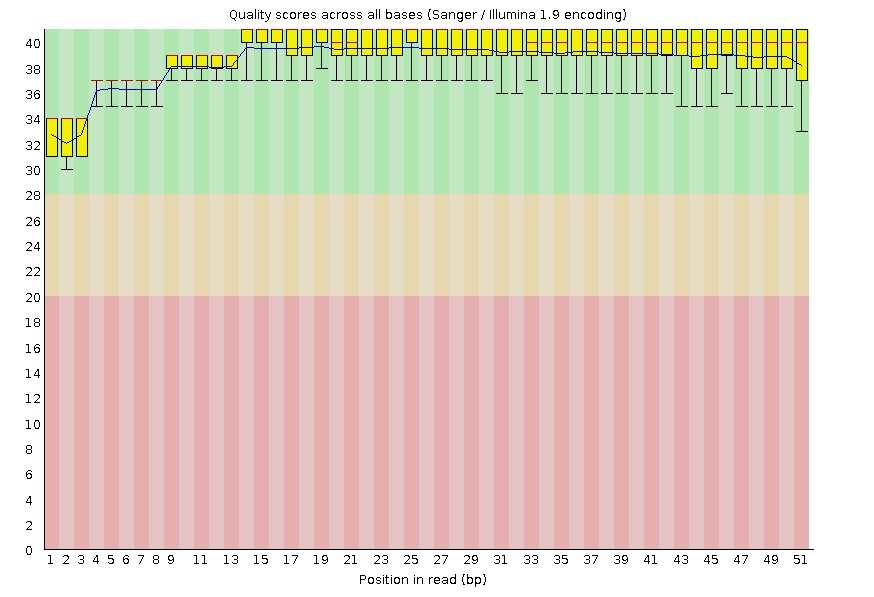
До чистки
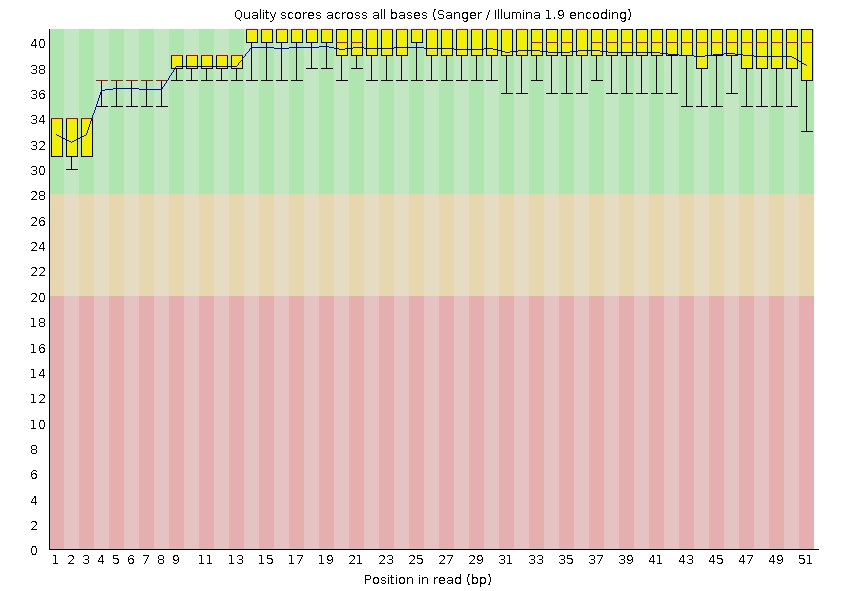
После чистки
Часть 2-3. Картирование и анализ чтений
Картирование чтений было произведено программой hisat2, экспортированной с помощью командыexport PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5
fastqc chr21.1.fastqОчистка чтений производилась с помощью программы Trimmomatic. Она была запущена с такими параметрами, чтобы были удалены чтения с длиной менее 50 нуклеотидов и с концов каждого прочтения были удалены нуклеотиды с качеством ниже 20.
java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr21.1.fastq chr21.1_trimm.fast TRAILING:20 MINLEN:50
| Число чтений до чистки | Число чтений после чистки |
|---|---|
| 11221 | 11158 |
export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5
| Команды | Описание |
|---|---|
hisat2-build chr21.fasta chr21_proc | Производит индексирование референсной последовательности |
hisat2 -x chr21_proc -U chr21_trimm.fastq --no-softclip>chr21.sam | Выравнивание прочтений и референса |
samtools view -b chr21.sam -o chr21.bam | Перевод в бинарный формат |
samtools sort chr21.bam -T text.txt -o chr21_sort.bam | Сортировка выравниваний по координате в референсе начала чтения |
samtools index chr21_sort.bam | Индексирование отсортированного выравнивания |
samtools idxstats chr21_sort.bam>res.txt | Запись числа откартированных чтений |
htseq-count -f bam -s no -m union -i gene_id chr21_sort.bam gencode.v19.chr_patch_hapl_scaff.annotation.gtf >> count_2.txtОпции запуска: -f формат bam или sam -s цепь прямая(yes) или обратная(no) -i атрибут gff -m режим обработки перекрытия ридов Результат работы программы: no_feature 408 ambiguous 0 too_low_aQual 0 not_aligned 281 alignment_not_unique 0
htseq-count -f bam -s no -m intersection-strict -i gene_id chr21_sort.bam gencode.v19.chr_patch_hapl_scaff.annotation.gtf >> count_3.txtРезультат запуска с опцией intersection-strict: ENSG00000156256.10 110 ENSG00000156261.8 9878 ENSG00000231125.2 21 Можно увидеть, что теперь уменьшилось число ридов на каждый ген, кроме последнего