Практикум 12
Сценарий cmp_msa для сравнения выравниваний
Был создан сценарий cmp_msa.py (скачивать сценарий следует с помощью командной строки: wget https://kodomo.fbb.msu.ru/~slavik123/term2/cmp_msa.py; для корретного отображения информации) на языке Python для того, чтобы проводить сравнение двух выравниваний, сделанных разными программами MSA. Сценарий использует векторный подход для сравнения выравниваний. Для того чтобы ознакомиться с инструкцией по работе и использованию данного сценария можно написать в командной строке, находясь в соответствующей директории, следующее:
Сравнение разных программ MSA
Поскольку на сегодняшний день нет абсолютно точной программы множественного выравнивания, то в качестве референсного выравнивания для проведения данного сравнительного анализа было взято одно из выравниваний из базы данных BAliBASE (ссылка на файл с этим выравниванием, это файл BB50014 из архива BAliBASE_R1-5.tar.gz с оффициального сайта BAliBASE). Выравнивание содержит 30 белков из разных организмов (дрожжи, нематоды, млекопитающие, птицы, рыбы), сами белки являются липазами, карбоксилэстеразами, ацетилхолинэстеразами и т.п., главное, что все они обладают эстеразной активностью (EC: 3.1.1) благодаря наличию крупного домена: Карбоксилэстеразный (КОэстераза) (PF00135). Данное выравнивание отражает эволюцию и гомологию различных эстераз у разных организмов.
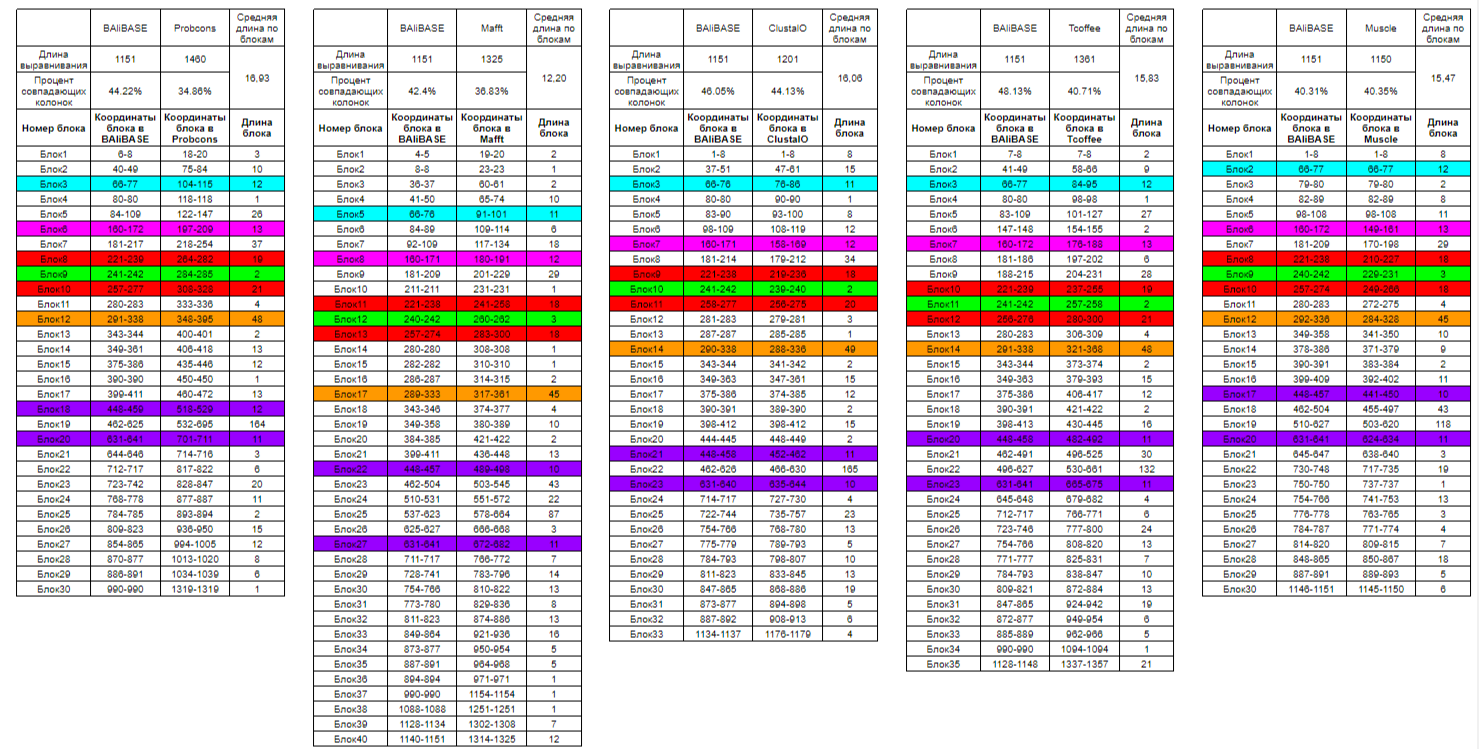
С референсным выравниванием сравнивались 5 программ множественного выравнивания, доступных в Jalview: Probcons, Mafft, ClustalO, Tcoffee, Muscle. Для сравнения результатов выравнивания был использован сценарий cmp_msa.py, описанный выше. Результаты представлены в Табл. 1 ниже:
Сами выравнивания проводились с помощью программы Jalview (ссылка на проект). Прилагается также архив из координат совпавших колонок для каждой программы (ссылка на архив).
Из таблицы видно в целом, что все программы еще далеки от совершенного идеала (процент совпадения везде ниже 50%), если разумеется принимать данные из BAliBASE как абсолютно точные, тем не менее это не означает, что они некорретные: при взгляде отдельно на выравнивания программ в общем и целом они достаточно хорошо похожи на референсное, основные высококонсервативные участки гомологии (позиции 40-110, 160-175, 220-240, 250-340, 350-460, 630-750, 780-860, 890-930 в референсном выравнивании) хорошо прослеживаются во всех выравниваниях и к тому же в рамках абсолютной точности и векторного подхода, учитывая огромный размер данных, те 30-45% процентов совпадения это очень качественный результат (в грубом приближении можно считать, что выравнивания совпадают на 70-85%).
Однако все эти программы используют разные алгоритмы для построения выравнивания, поэтому какой-то окажется более точным. Основные участки гомологии в общем-то различаются довольно мало у разных программ, наиболее "субъективно" программы выровняли следующие позиции: N-конец (позиции 1-30 в референсе), крупный общий индель 460-625 в референсе (вызванный по всей видимости многократной дупликацией короткого участка у белка ACES_CHICK), концевые участки белка (позиции с 930 и до конца в референсе). Для начала обратим внимание на те блоки, которые наиболее точно совпали во всех 5 программах:
- Голубой цвет в таблице соответствует консервативному блоку 66-77 в референсе.
- Пурпурный цвет в таблице соответствует консервативному блоку 160-172 в референсе.
- Красным показаны консервативные позиции 221-239 и 257-277 в референсе, разделенные возникшей у группы 3 белков G-петлей (ей соответствует зеленый цвет).
- Оранжевый цвет в таблице соответствует консервативному блоку 290-340 в референсе.
- Фиолетовые цвета показывают консервативные позиции до и после крупного инделя (448-459 и 631-641 соответственно в референсе).
Таким образом, выравнивания хорошо совпадают на протяжении всего КОэстеразного домена (главный консервативный участок для всех белков, подтверждающий их гомологию). Подобный анализ совпадения блоков во всех программах с разным алгоритмом позволяет сделать выводы о корректности и правильности выравнивания на этих совпадающих участках, это один из вариантов увидеть выравнивание, наиболее близкое к эволюционному, на определенном участке у белков.
Наиболее точной программой в данном случае оказался ClustalO (он же ClustalΩ) по суммарному вкладу глобальных характеристик (наивысший процент совпадающих колонок – 44.13%, длина выравнивания близка к референсу, высокая средняя длина совпадающих блоков, а также их большое число), именна эта же программа абсолютно точно выровняла тот самый крупный индель (Блок22 в 3 таблице). Также довольно точной оказалась программа Muscle (длина ее выравнивания практически совпала с референсом, остальные показатели также высоки), среднее положение заняла программа Tcoffee, наименее точными оказались программы Mafft (низкий процент совпадения, огромное число совпадающих блоков, но очень мелких по длине (в частности единичных), большая длина выравнивания по сравнению с референсом) и Probcons (низкий процент совпадения и очень большая длина выравнивания). По всей видимости алгоритмы Probcons и Mafft создали большое число эволюционно неверных гэпов, из-за чего и большие длины выравниваний и низкий процент совпадения.
Подводя итоги можно утверждать, что все программы в той или иной степени довольно хорошо выравнивают консервативные блоки (такие как крупные домены), однако плохо справляются с выравниванием тех участков, где и можно наблюдать действие эволюционного отбора (участки различий последовательностей): так позициям 1-30 и 900-1100 (в референсе) практически нет совпадений ни в одном из 5 случаев (а именно эти позиции отражают расхождение белков с эстеразной активностью у разных организмов).
3D-выравнивание
Для проведения 3D выравнивания было выбрано следующее семейство доменов из Pfam: Трипсин (PF00089). Выравнивались следующие три белка:
- 1GDQ – трипсин из Fusarium oxysporum.
- 1QQU – трипсин из Sus scrofa.
- 1OSS – трипсин из Streptomyces griseus.
Пространственное выравнивание осуществлялось с помощью Pairwise Structure Alignment (в качестве референса была взята структура 1GDQ) на сайте PDB алгоритмом TM-align (результаты представлены в Табл. 2 и на Рис. 1), обычное же выравнивание производилось с помощью программы ClustalO в Jalview (ссылка на проект, там же и 3D-выравнивание из PDB).
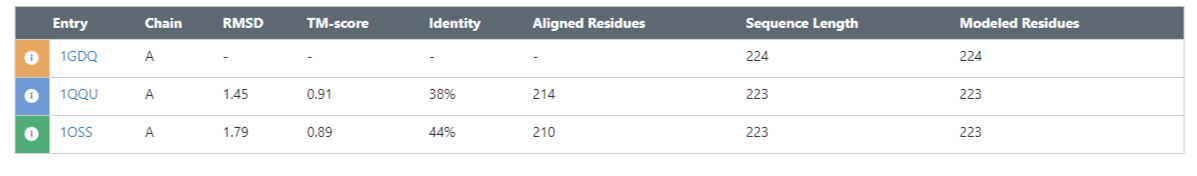
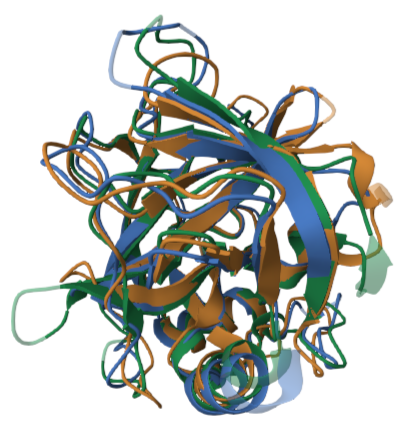
К сожалению, программа на PDB не способна построить 3D-выравнивание сразу для всех трех, поэтому сравнить результат статистически с помощью сценария cmp_msa как в прошлом пункте не удастся (а проводить выравнивание двух выравниваний 3D с помощью программы MSA не имеет смысла, поскольку получаются на 100% одинаковые результаты, это явно искажает пространственное исходное выравнивание). Тем не менее оба выравнивания позволяют сделать один и тот же вывод про эти белки: из выравниваний видно, что трипсины довольно сильно отличаются, есть лишь небольшое количество высококонсервативных участков (2-4, 26-30, 36-43, 124-126, 174-186, 200-220 в выравнивании ClustalO), которые соответствуют хорошо совмещенным вторичным структурам на 3D-выравнивании. Остальные позиции не являются консервативными и соответствуют совершенно несовместимым многочисленными петлям и изгибам между теми самыми вторичными структурами (смотри Рис. 1). По всей видимости образование разных петель и изгибов с разной топологией и геометрией потребовалось для правильной ориентации сайтов и участков домена трипсина (это и есть те самые консервативные позиции), учитывая разную биологию и экологию всех трех организмов.
Описание программы ClustalΩ
Clustal – это компьютерная программа, необходимая для построения множественного выравнивания нескольких последовательностей (как белковых, так и нуклеиновых). Данная программа была создана в 1988 году Десмондом Джерардом Хиггинсом. На протяжении своего существования и по сей день программа улучшалась, последняя ее версия носит название ClustalΩ (написана на языках C и C++).
Программа принимает на вход (нужно как минимум 3 последовательности) широкий спектр форматов: NBRF/PIR, FASTA, EMBL/Swiss-Prot, Clustal, GCC/MSF, GCG9 RSF и GDE. Формат выходного файла может быть следующим: Clustal, NBRF/PIR, GCG/MSF, PHYLIP, GDE, NEXUS.
1.Алгоритм
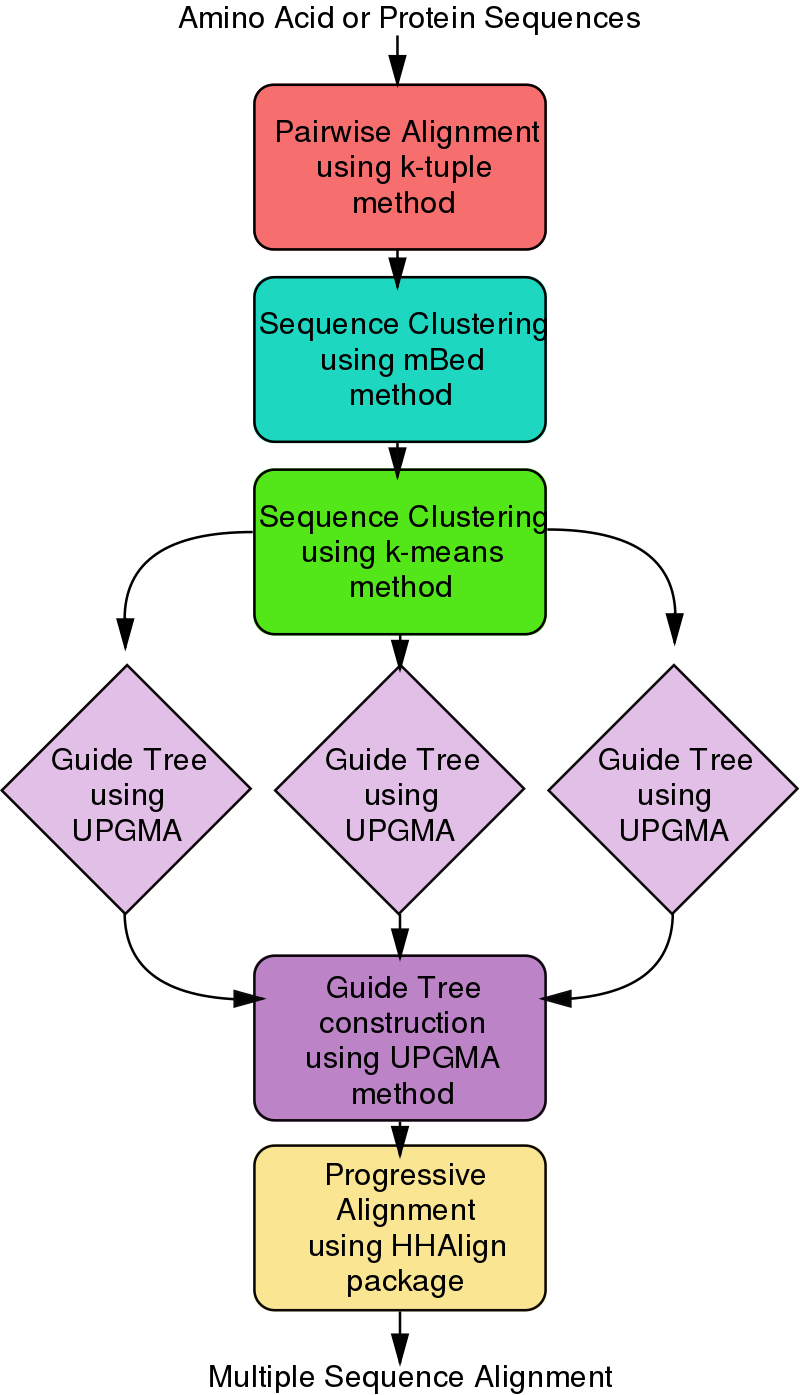
Алгоритм данной программы включает 5 шагов (Рис. 1):
- Строятся всевозможные попарные выравнивания с помощью метода k-кортежей (более эффективный чем динамическое программирование).
- С помощью модифицированного mBed метода расчитываются попарные расстояния.
- Последовательности кластеризуются методом k-средних из кластерного анализа.
- Для каждого кластера строится свое локальное дерево методом UPGMA (используя ранее расчитанные расстояния), которы затем объединяются в одно единое глобальное направляющее дерево.
- На основе этого глобального дерева строится само множественное выравнивание с помощью пакета HHAlign из HH-Suite, используя два профиля HMM.
2.Результаты
Временная сложность точного расчета оптимального множественного выравнивания для N последовательностей длины L составляет O(LN), однако благодаря модифицированному методу mBed временная сложность сводится к O(N·log(N)), что значительно сокращает время вычисления и требования к памяти. ClustalΩ превосходит многие другие алгоритмы по времени, точности и памяти, он способен запускать более 100 000 последовательностей за несколько часов (это достигается за счет модифицированного метода mBed и пакета HHAlign). По скорости в тесте эффективности ClustalΩ уступал лишь Kalign.
3.Источники
- Clustal (Электронный ресурс): Wikipedia. The Free Encyclopedia. Режим доступа: https://en.wikipedia.org/wiki/Clustal (дата обращения 09.05.2024).