Практикум 8
Задание 1
В данном задании необходимо было найти в сборке генома выбранного организма (в моем случае это роющая актиния Nematostella vectensis) короткий контиг и провести для него поиск BLAST.
Для поиска участков подходящей длины в скачанном с сайта NCBI файле genomic.gbff, содержащем аннотированные последовательности генома, я написал скрипт на python (notebook, с программами данного практикума доступен по ссылке). Однако оказалось, что в сборке отсутствуют контиги длиной в 1000 - 10000 п. н., содержащие CDS. Поэтому я повысил верхний порог по длине до 15000.
Из выдачи скрипта я выбрал участок с идентификатором NW_026019263 длиной в 12138 п. н., содержащий одну CDS, соответствующую белку rho-related GTP-binding protein RhoB (protein id XP_048579143.1). Для получения фрагмента подходящей длины я решил вырезать из найденного участка ген данного белка (с 7783-его по 11716-ый нуклеотидный остаток в представленной в файле цепи, Рис. 1).
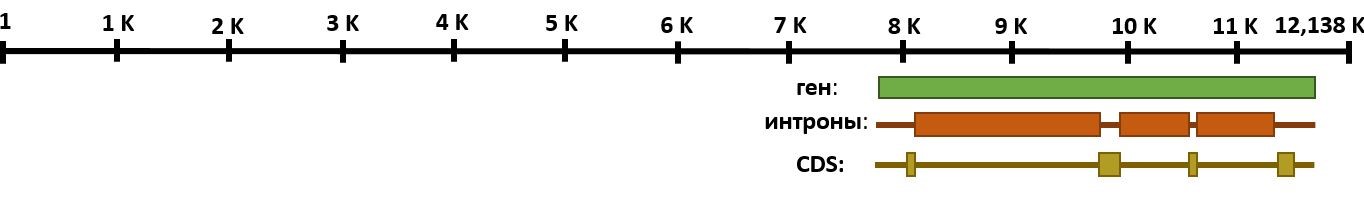
Файл с последовательностью полученного фрагмента в формате FASTA доступен по ссылке. Соответствующий ему ген содержит 4 экзона и кодирует белок RhoB - ГТФазу, гомолог которой у человека участвует в миграции клеток, внутриклеточном транспорте везикул, репарации ДНК, апоптозе и ряде других процессов.
Далее, пользуясь сайтом NCBI, для полученного фрагмента я запустил различные алгаритмы BLAST: blastn, megablast, blastx и tblastx. Изменяемые параметры и количество находок в выдаче от каждого алгоритма представлены в таблице ниже (Табл. 1).
| Метод BLAST | Word size | Порог по E-value | Вес Match/Mismatch |
Штрафы за открытие/продление гэпа |
Максимальный вес лучшей находки* |
E-value лучшей находки |
Число находок |
|---|---|---|---|---|---|---|---|
| blastn | 11 | 0.05 | 2/-3 | -5/-2 | 151 | 1e-30 | 4315 |
| megablast | 20 | 0.05 | 1/-2 | Линейные | 97.1 | 2e-14 | 116 |
| blastx | 6 | 0.05 | матрица BLOSUM62 |
-11/-1 | 55.8 | 3e-04 | 184 |
| tblastx | 3 | 0.01 | матрица BLOSUM62 |
-11/-1 | - | - | - |
* Имеется в виду лучшая по максимальному весу находка.
Перед запуском BLAST я исключил из поиска таксон Cnidaria (тип Стрекающие, NCBI taxid 6073). Для каждого из методов был использован выставленный по умолчанию порог на E-value (0.05), максимольный размер выдачи быз взят за 500 (5000 в случае blastn).
Метод megablast работал быстрее всех, однако из-за большого размера "слова" и линейных штрафов за гэпы по нему нашлось наименьшее число последовательностей.
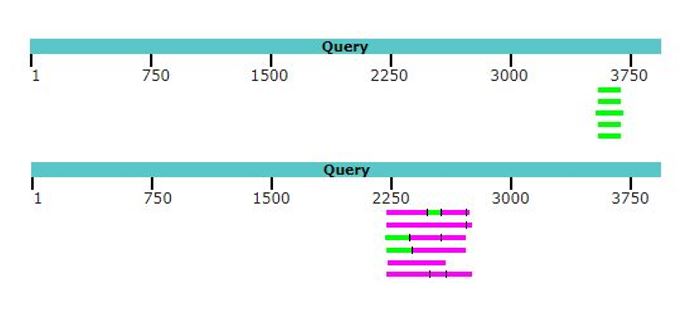
На первый взгляд, наилучший результат был получен с помощью метода blastn. Однако в этом случае (как и в случае megablast) общий вес находки часто в несколько раз превышал максимальный, а найденная последовательность сравнительно часто была локализована на половой хромосоме. На самом деле, большинство находок представляло собой выравнивания с участком запроса в примерной области 2230 - 2440 (Рис. 2), целиком принадлежащей второму интрону гена. Найденные же последовательности почти целиком состояли из четырехбуквенных повторов (Рис. 3).
Действительно значимый результат дал метод blastx. В этот раз найденные последовательности в среднем соответствовали нуклеотидным остаткам 3535 - 3680 запроса (Рис. 2). Исходя из координат интронов в гене, этот участок вплотную прилежит к третьему интрону и почти полностью входит в четвертый экзон. Что не менее важно, большую часть выдачи составляли Rho-белки и другие ГТФазы. Однако данный метод не получилось запустить при длине слова меньше 6.
Запустить метод tblastx на сайте NCBI не получилось даже с минимальными настройками и после ограничения поиска "сверху" таксоном Metazoa. Поэтому в данном случае я решил использовать сайт EBI. Запрос обрабатывался несколько дней, однако результатов не дал (сообщение "Job failed").
Таким образом, blastn и megablast стоит использовать только в том случае, если в последовательности запроса нет "лишних" участков большого размера (например, при поиске гомологов по последовательности мРНК или гена бактерии). При этом megablast работает намного быстрее, но пропускает большинство потенциальных находок в случае их низкой степени схожести с последовательностями банка данных. Использование этого метода имеет смысл при работе с последовательностями близкородственных организмов или с сильно консервативными последовательностями (например, рРНК). blastx работает дольше blastn, но позволяет использовать в качестве запроса последовательности, содержащие много длинных некодирующих участков. В частности, в данном случае (поиск гомологов гена эукариотического организма) этот метод оказался наиболее оптимальным.
Задание 2
Для выполнения данного задания я решил пользоваться программами, установленными на сервере kodomo.
Для поиска в геноме Nematostella vectensis последовательностей, гомологичных 16S рРНК и 23S рРНК E. coli (входят в состав малой и большой субъединиц рибосомы соответственно), я загрузил соответствующие файлы на сервер (файл rRNA_ecoli.txt я разделил на 16S_rRNA_ecoli.fasta и 23S_rRNA_ecoli.fasta) и проиндексировал используемый в поиске BLAST банк данных (геном актинии):
makeblastdb -in GCF_932526225.1_jaNemVect1.1_genomic.fna -dbtype nucl -out db.fasta
Хотя последовательности рРНК являются достаточно консервативными, в данном случае речь идет о поиске далеких гомологов в небольшом банке данных. Поэтому я решил использовать более чувствительный алгоритм blastn. Были выполнены следующие команды:
blastn -task blastn -query 16S_rRNA_ecoli.fasta -db db.fasta -out 16S.out -evalue 0.05 -word_size 7 -outfmt 7
blastn -task blastn -query 23S_rRNA_ecoli.fasta -db db.fasta -out 23S.out -evalue 0.05 -word_size 7 -outfmt 7
При поиске я использовал порог на E-value 0.05, длину затравки 7, вес Match/Mismatch 2/-3 и штрафы за гэпы -5/-2. Файлы с примерами выдачи (для каждой найденной последовательности локального банка данных приведено только лучшее выравнивание) доступны по ссылкам:
По обоим запросам было получено очень большое число находок: 435 для 16S и 189 для 23S рРНК, причем похожие участки были найденны на большинстве хромосом. Я решил проанализировать только лучшие и худшие из них.
Для 23S рРНК лучшая находка имела E-value 2е-16 и соответствовала фрагменту гена, с которого транскрибируется 28S рРНК, локализованного на участке NW_026019260 неопределенного положения. Худшее выравнивание имело E-value 0,03. Найденная последовательность является частью девятой хромосомы и расположена на сравнительно небольшом расстоянии от гена 5S рРНК.
В выдаче от поиска по 16S рРНК и лучшая, и худшая находки соответствовали участкам рДНК, кодирующим 18S рРНК. При этом последовательность лучшей находки принадлежит четверьой хромосоме, а последовательность худшей - уже упомянутому участку NW_026019260.