Практикум 6
Задание 1
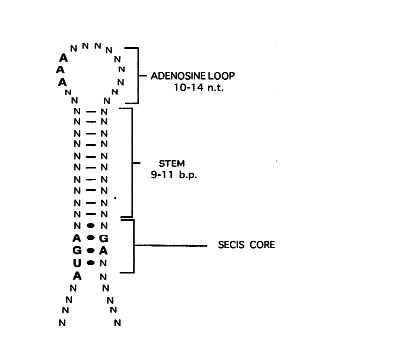
В качестве сигнала для описания я выбрал SECIS-элемент, отвечающий за котрансляционное встраивание в синтезируемый белок нетрадиционной аминокислоты селеноцистеина (Sec). Эта аминокислота иногда встречается в активных центрах белков, обладающих антиоксидантной активностью. Так как у нее отсутствует свой собственный кодон, особая селеноцистеиновая тРНК взаимодействует с кодоном UGA, на котором в норме происходит остановка трансляции. Необходимый "контекст" при этом обеспечивает специальная шпилька (SECIS-элемент, Рис. 1), с которой связываются белки, привлекающие Sec-тРНК и обеспечивающие паузу в работе рибосомы.
Данный механизм обнаружен в каждом из трех доменов живых организмов: у бактерий, архей и эукариот. Однако его детали, такие как консенсусная последовательность SECIS-элементов, их количество и расположение в мРНК, а также участвующие в процессе белки, заметно отличаются (Grundner-Culemann, E. et al., 1999).
Описываемый сигнал является достаточно эффективным, так как его игнорирование приводило бы к преждевременной терминации трансляции с образованием неполного белка, лишенного рабочего активного центра.
Two distinct SECIS structures capable of directing selenocysteine incorporation in eukaryotes
Задание 2
Для выполнения этого и последующих заданий был написан ряд программ на языке python:
В качестве сигнала для описания я выбрал последовательность Шайна-Далгарно (SD) бакттерии Escherichia coli штамма K-12 подштамма MG1655. Эта последовательность расположена в 5'-некодирующем участке мРНК и участвует в инициации трансляции у прокариот, гибридизуясь с участком 16S рРНК.
С сайта NCBI была скачана таблица локальных особенностей генома выбранного штамма (RefSeq ID GCF_000005845.2, референсный) в формате gbff. Для получения участков, содержащих последовательность Шайна-Далгарно, были использованы консольные команды пакета EMBOSS:
extractfeat "genomic.gbff" "ecoli_cds.fasta" -type CDS -before 20 -filter
seqret "ecoli_cds.fasta[1:23]" "ecoli_sd.fasta" -filter

Число нуклеотидов до старт-кодона было изначально взято за 20. Fasta-файл, содержащий полученные последовательности, выравненные по старт-кодонам, был открыт в Jalview. С помощью функций программы были выделены участки, соответствующие консенсусной последовательности SD Escherichia coli - 5'-GGAGG-3' (Saito, K. et al., 2020). Оказалось, что расстояние между собственно сигналом и старт-кодоном варьирует в достаточно широких пределах, хотя в выравнивании все же есть несколько достаточно консервативных колонок (Рис. 2). Далее имело бы смысл выравнять фрагменты генов по последовательностям SD, однако в таком случае пришлось бы исключить из выравнивания старт-кодоны. В итоге я решил работать с последовательностями SD как с пурин-богатыми участками на определенном расстоянии от старт-кодона. С учетом положения выделенных участков и аннотации выравнивания была выбрана условная точка начала сигнала, расположенная на 11 нуклеотидов выше ATG. Из полученных ранее последовательностей были вырезаны соответствующие сегменты (от начала SD до старт-кодона).
1735 (~40%) из полученных последовательностей были определены в тренировочную выборку, а 2602 оставшиеся - в тестовую. Группа для негативного контроля была построена из 2602 случайно выбранных из того же генома участков, каждый из которых содержал нестартовый ATG и 11 предшествующих ему нуклеотидных остатков.
Далее по материалу обучения была построена позиционная весовая матрица (Табл. 1). Значения pseudocounts для каждого из остатков были взяты за 0.1, а для подсчета отношений правдоподобия использовался GC-состав генома выбранной бактерии, равный 0.505.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 0.44 | 0.23 | 0.20 | 0.33 | 0.40 | 0.28 | 0.28 | 0.41 | 0.53 | 0.06 | 0.11 | 1.28 | -4.43 | -4.93 |
| T | -0.76 | -0.72 | -0.93 | -0.63 | -0.27 | 0.03 | 0.10 | 0.03 | -0.30 | 0.30 | 0.22 | -2.59 | 1.39 | -4.25 |
| G | 0.44 | 0.68 | 0.77 | 0.58 | 0.29 | 0.09 | -0.13 | -0.39 | -0.14 | -0.63 | -0.48 | -1.07 | -4.67 | 1.37 |
| C | -0.85 | -1.29 | -1.52 | -1.25 | -0.88 | -0.57 | -0.36 | -0.24 | -0.36 | 0.06 | 0.02 | -5.34 | -5.99 | -4.45 |
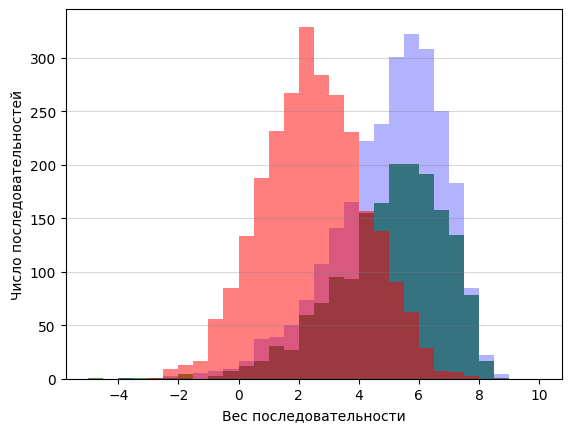
После этого для всех полученных групп с использованием построенной матрицы были рассчитаны веса входящих в них последовательностей. Результаты были представлены в виде гистограммы (Рис. 3).
| Train | Test | Negative control | |
|---|---|---|---|
| сигнал(+) | 1300 (75%) | 1935 (74%) | 492 (19%) |
| сигнал(-) | 435 (25%) | 667 (26%) | 2110 (81%) |
Порог по весу для разделения находок на правильные и ложные был взят за 4. Результаты разделения представлены в таблице (Табл. 2.). По гистограмме явно видно, что параметры распределения весов элементов тренировочной и тестовой выборок совпадают. Это вполне ожидаемо, так как они были взяты из одной генеральной совокупности. Распределение же для материала отрицательного контроля заметно смещено влево. Несмотря на это, оно сильно пересекается с распределениями для первых двух групп, что отразилось и на количестве последовательностей, прошедших порог по весу в каждой из выборок (в данном случае невозможно подобрать порог, который обеспечил бы эфффективное разделение).
Причина неточности разделения заключается, вероятно, в вариативности длины участка между последовательностью SD и старт-кодоном. Однако, стоит также отметить, что сила исследуемого сигнала (и его схожесть с консенсусом) в разных генах сильно различается, так как наличие последовательности SD не является строго обязательным условием для инициации трансляции (Saito, K. et al., 2020).
Задание 3
По материалу обучения были построены матрица информационного содержания (Табл. 3.) и Logo-диаграмма (Рис. 4) последовательности Шайна-Далгарно Escherichia coli.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 0.24 | 0.11 | 0.09 | 0.17 | 0.21 | 0.13 | 0.13 | 0.22 | 0.32 | 0.02 | 0.04 | 1.66 | -0.02 | -0.01 |
| T | -0.13 | -0.12 | -0.13 | -0.12 | -0.07 | 0.01 | 0.04 | 0.01 | -0.08 | 0.14 | 0.10 | -0.07 | 1.99 | -0.02 |
| G | 0.25 | 0.48 | 0.60 | 0.37 | 0.14 | 0.03 | -0.04 | -0.10 | -0.04 | -0.12 | -0.11 | -0.13 | -0.02 | 1.96 |
| C | -0.13 | -0.13 | -0.12 | -0.13 | -0.13 | -0.12 | -0.09 | -0.07 | -0.09 | 0.02 | 0.01 | -0.01 | -0.01 | -0.02 |
| IC(j) | 0.23 | 0.34 | 0.44 | 0.29 | 0.15 | 0.06 | 0.04 | 0.07 | 0.11 | 0.07 | 0.04 | 1.44 | 1.96 | 1.91 |
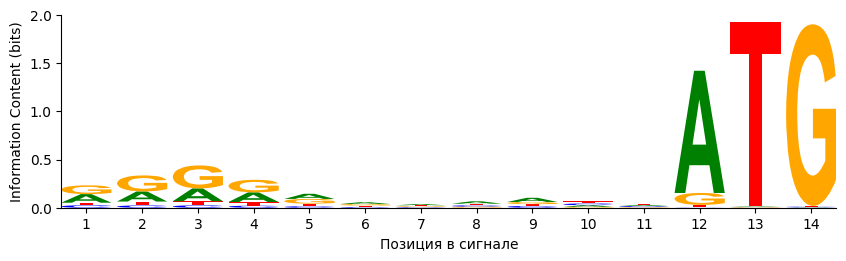
На лого-диаграмме хорошо виден пурин-богатый участок в позициях с 1 по 5. Достаточно большая доля аденина характкрна также для позиции 9.
Задание 4
Для выполнения данного задания я использовал геном все того же штамма Escherichia coli str. K-12 substr. MG1655. Число сайтов 5'-GAATTC-3' в геноме составило 646.
Последовательность данного участка является паллиндромом, поэтому число сайтов на прямой цепи всегда будет равно их количеству на обратной. Все дальнейшие расчеты проведены для одной цепи ДНК.
Для расчета вероятности (ожидаемой) появления участка GAATTC начиная с определенной позиции в геноме частоты составляющих сайт нуклеотидов (с учетом GC-состава) были перемножены между собой. Матожидание числа встреч можно получить, домножив полученное значение на длину генома (4641652 п.н.).
p = 0.25252 × 0.24754 = 2.39 × 10-4
E = p × 4641652 ≈ 1110.44
Найденное значение составило примерно 1110, что почти в два раза больше реального количества таких участков в геноме. Для оценки значимости выявленного различия был использован обычный Z-тест. Нулевая гипотеза в данном случае заключается в том, что среднее число встреч сайта GAATTC равно 1110.44. В случае верности альтернативной гипотезы математическое ожидание мельше вышеупомянутой величины.
H0: μ = 1110.44;
Ha: μ < 1110.44
Случайная величина X, равная количеству сайтов в геноме, имеет биномиальное распределение, причем из-за достаточно большой длины генома ее можно аппроксимировать нормальным распределением (по Центральной предельной теореме).
X ∼ Bin(46411652, 2.39 × 10-4) ≈ N(np, √(npq))
X ∼ N(1110.44, 33.3193)
P-value = P(X ≤ 646) = P(Z ≤ (646 - 1110.44)/33.3193) =
= P(Z ≤ -13.94) = 1.8 × 10-44
Значение параметра P-value составило примерно 1.8 × 10-44, что позволяет отвергнуть нулевую гипотезу при почти любом уровне значимости. Таким образом, в геноме Escherichia coli участок с последовательностью GAATTC по определенной причине действидельно встречается реже ожидаемого.
Литература
- Grundner-Culemann, E., Martin, G. W., 3rd, Harney, J. W., & Berry, M. J. (1999). Two distinct SECIS structures capable of directing selenocysteine incorporation in eukaryotes. RNA (New York, N.Y.), 5(5), 625–635. https://doi.org/10.1017/s1355838299981542
- Saito, K., Green, R., & Buskirk, A. R. (2020). Translational initiation in E. coli occurs at the correct sites genome-wide in the absence of mRNA-rRNA base-pairing. eLife, 9, e55002. https://doi.org/10.7554/eLife.55002