Практикум 4
В этом практикуме для предсказания структур белков по их последовательностям использовалась нейросеть AlphaFold2, установленная в Google Colab (ColabFold v1.5.5).Задание D8
В данном задании нужно было оценить влияние N-концевой делеции в белке на структуру, предсказанную AlphaFold. Для этого использовались две последовательности, принадлежащие (судя по выдаче blastp) D-допахром декарбоксилазе. Одна из них соответствует целому ферменту, тогда как в другой отсутствуют первые 15 аминокислотных остатков.
Для обоих ферментов были предсказаны структуры. Судя по метрикам из выдачи AlphaFold, качество предсказания является достаточно хорошим (Рис. 1). Важно также отметить, что в данном случае оно практически не зависит от наличия делеции. В обоих случаях для всех пяти моделей значение pIDDT (predicted local distance difference test), дающее оценку сходства с реальной структурой, не опускается ниже 80 (из 100). В то же время метрика PAE (predicted aligned error), предсказывающая неточность взаимного расположения разных участков модели, для большинства пар остатков принимает значения, близкие к 0.
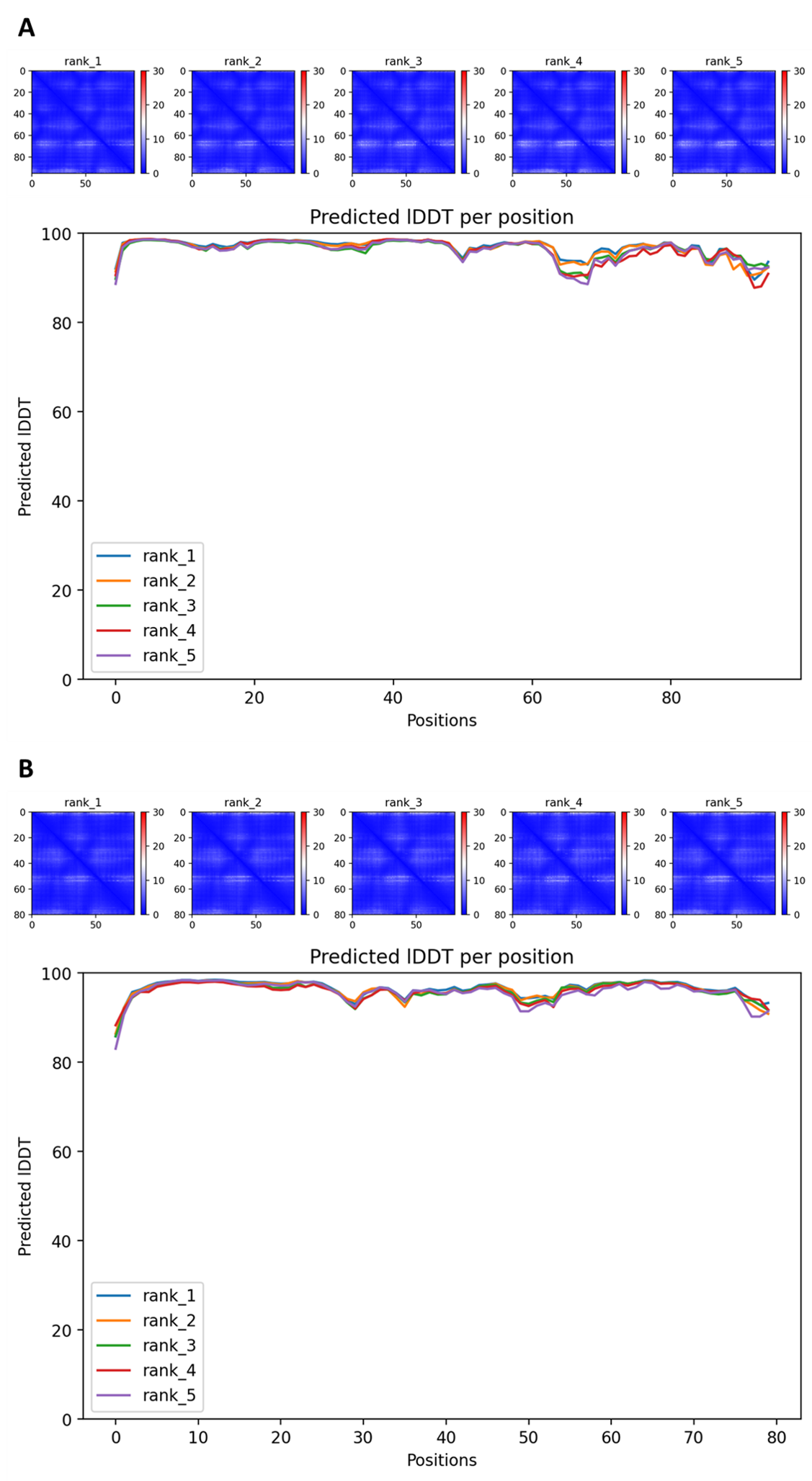
Несмотря на хорошие значения метрик, при близком рассмотрении самих структур становится понятно, что фермент с делецией, скорее всего, был смоделирован неправильно. В предсказанной структуре целого белка присутствуют 2 α-спирали и небольшой антипараллельный β-лист из 3 тяжей. При этом делетированный участок соответствует центральному тяжу β-листа. Стоило ожидать, что в белке с делецией фрагменты, соответствующие остальным тяжам, примут другую структуру, так как их β-листовая конформация не будет стабилизирована водородными связями с центральным тяжом. Однако конформация этих остатков в двух полученных структурах практически не отличается (Рис. 2).
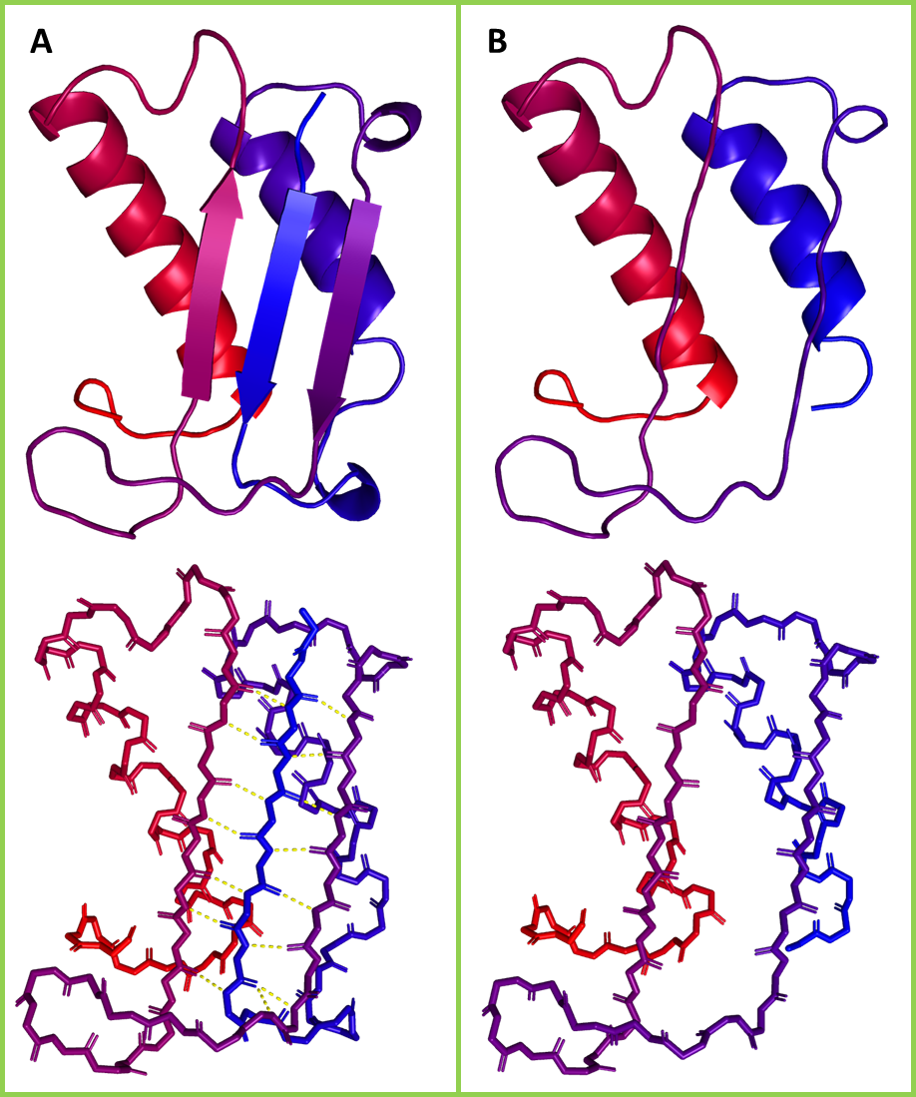
Вероятно, эта проблема связана с тем, что AlphaFold2 лишь предсказывает конечный результат фолдинга на основе имеющихся данных о структурах других белков. Она также может являться следствием переобучения. Полученная структура достаточно близка к известным, из-за чего она выбирается как удачная, однако она лишена важного участка, без которого правильный фолдинг становится невозможным. Скорее всего, похожий результат можно ожидать и для других делеций в белках с популярной архитектурой. Данные предположения можно проверить путем предсказания и (в качестве контроля) экспериментального получения подобных структур. Помимо экспериментальных структур для сравнения можно также использовать модели, полученные из общих соображений (ab initio) методами, не опирающимися на сходство с другими белками.
Задание A4
Одной из слабых сторон нейросети AlphaFold является ее способность к предсказанию структур фибрилляярных белков. Среди них особое место занимают амилоиды — агрегаты с β-листовой структурой, образующиеся в результате неправильного фолдинга белков клетки. Их отложение в различных тканях и органах нарушает их правильное функционирование и может приводить к развитию различных заболеваний (амилоидозов) [1]. Полимерная природа амилоидов и высокая степень полиморфизма в их структурах осложняют их исследование с использованием классических методов кристаллографии [2]. В связи с этим проблема точного предсказания структуры этих белков является достаточно актуальной.
Цель данного задания — сравнить полученную экспериментально структуру белка-амилоида со структурами, предсказанными с помощью AlphaFold.
Для поиска структур в базе данных PDB для выбранной последовательности использовалась программа BLAST (ее вариант blastp), установленная на сервере NCBI. Выдача blastp содержала несколько хороших находок, лучшая из которых (PDB ID 2NNT) полностью соответствовала последовательности запроса. Эта структура принадлежит амидоиду, образующемуся при агрегации WW2-доменов нескольких молекул транскрипционного активатора CA150 человека. В нативной конформации этот участок образует небольшой β-лист из 3 тяжей, тогда как в структуре амилоида он принимает форму "шпильки" (Рис. 3) [3].
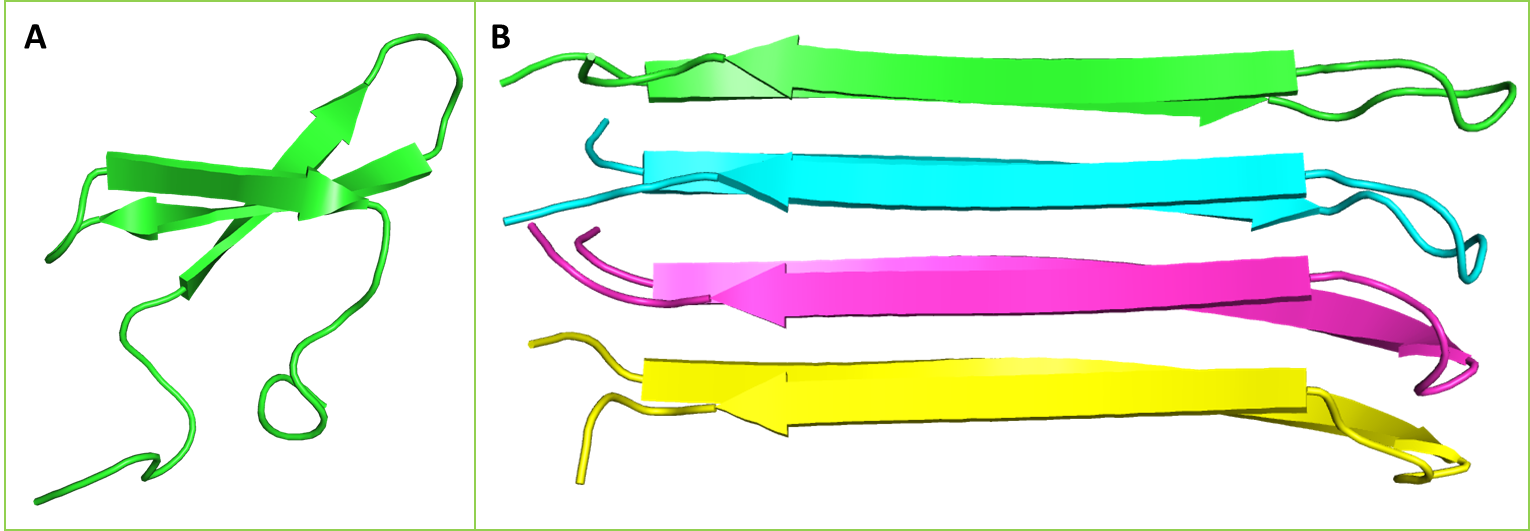
Для 2NNT были предсказаны структуры мономера и комплексов из 5 и 10 цепей (Рис. 4). Если для единственной цепи значение pIDDT опускалось ниже 60 только для подвижного N-конца молекулы, то в случае обоих комплексов оно было достаточно низким для всех остатков модели. Это говорит о низкой уверенности в предсказанной структуре мультимера. Если же сравнить полученные модели с двумя альтернативными структурами исследуемого белка (Рис. 3), то можно заметить, что выдача AlphaFold соответствует нативной конформации WW2-домена, а не образуемому им амилоиду.
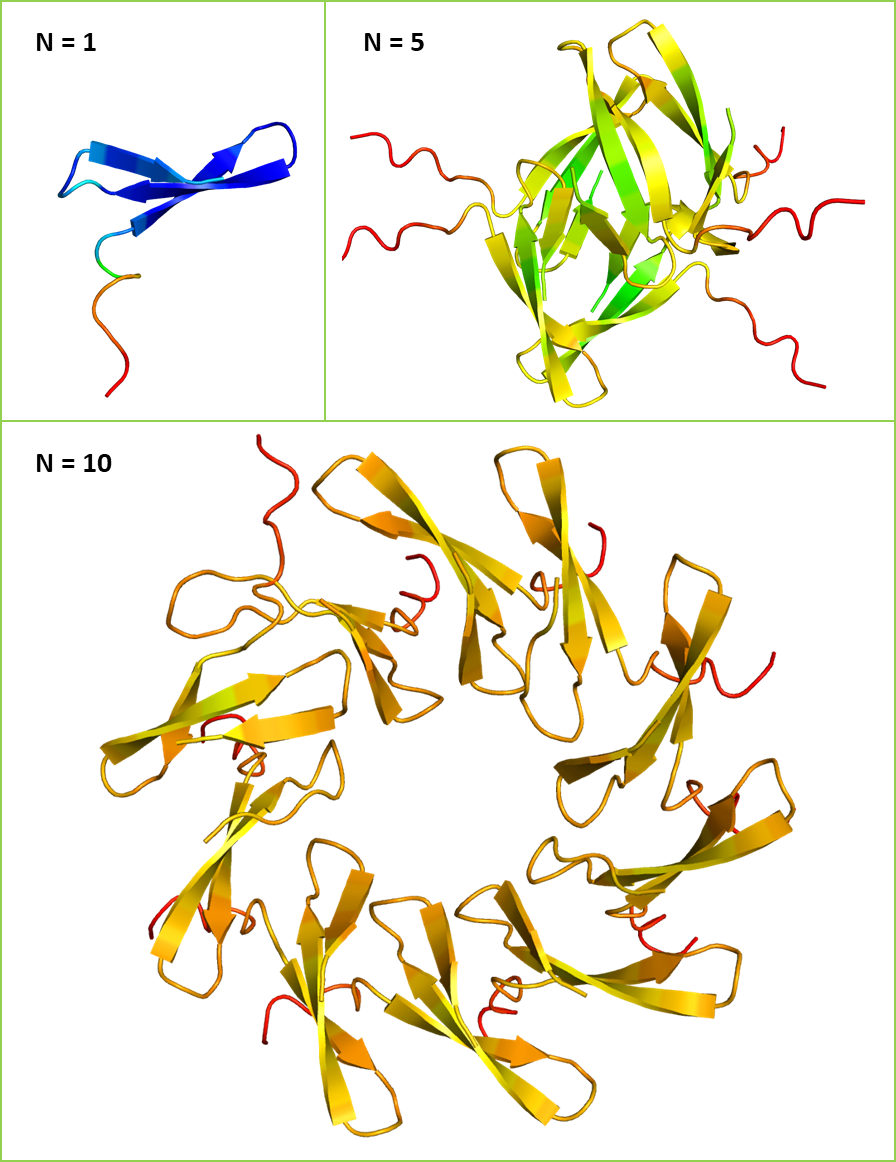
Судя по всему, в данном случае AlphaFold выдала неправильный результат из-за наличия у белка сильно отличающихся альтернативных конформаций. Вероятно, эта проблема также связана с тем, что нейросеть в целом лучше справляется с предсказанием структуры глобулярных белков и имеет "сдвиг" в сторону соответствующих архитектур. В связи с этим без дополнительных модификаций AlphaFold, скорее всего, плохо подходит для точного предсказания структуры амилоидов.
Ссылка на сессию pymol для Рис. 3
Ссылка на сессию pymol для Рис. 4
Источники
- Sunde, M., Serpell, L. C., Bartlam, M., Fraser, P. E., Pepys, M. B., & Blake, C. C. (1997). Common core structure of amyloid fibrils by synchrotron X-ray diffraction. Journal of molecular biology, 273(3), 729–739.
- Morris, K. L., & Serpell, L. C. (2012). X-ray fibre diffraction studies of amyloid fibrils. Methods in molecular biology (Clifton, N.J.), 849, 121–135.
- Ferguson, N., Becker, J., Tidow, H., Tremmel, S., Sharpe, T. D., Krause, G., Flinders, J., Petrovich, M., Berriman, J., Oschkinat, H., & Fersht, A. R. (2006). General structural motifs of amyloid protofilaments. Proceedings of the National Academy of Sciences of the United States of America, 103(44), 16248–16253.