Практикум 6
В этом практикуме я работал со двумя структурами: 6OQH и 2F1C. Они принадлежат белка OmpG E. coli, однако первая была получена методом ЯМР, а вторая — методом РСА. OmpG — белок со структурой бета-бочонка во внешней мембране E. coli. Он представляет собой потенциальную альтернативу для олигомерных белковых нанопор, используемых для секвенирования нуклеиновых кислот и измерения динамики фолдинга белков. Проблема с использованием OmpG заключается в наличии в его структуре между участками бета-тяжей подвижных петель, которые ограничивают возможность его применения для подобных задач.
Структура 2F1C имеет разрешение 2.30 Å. Запись 6OQH включает 10 из 200 рассчитанных конформеров с самой низкой энергией.
Задание 1
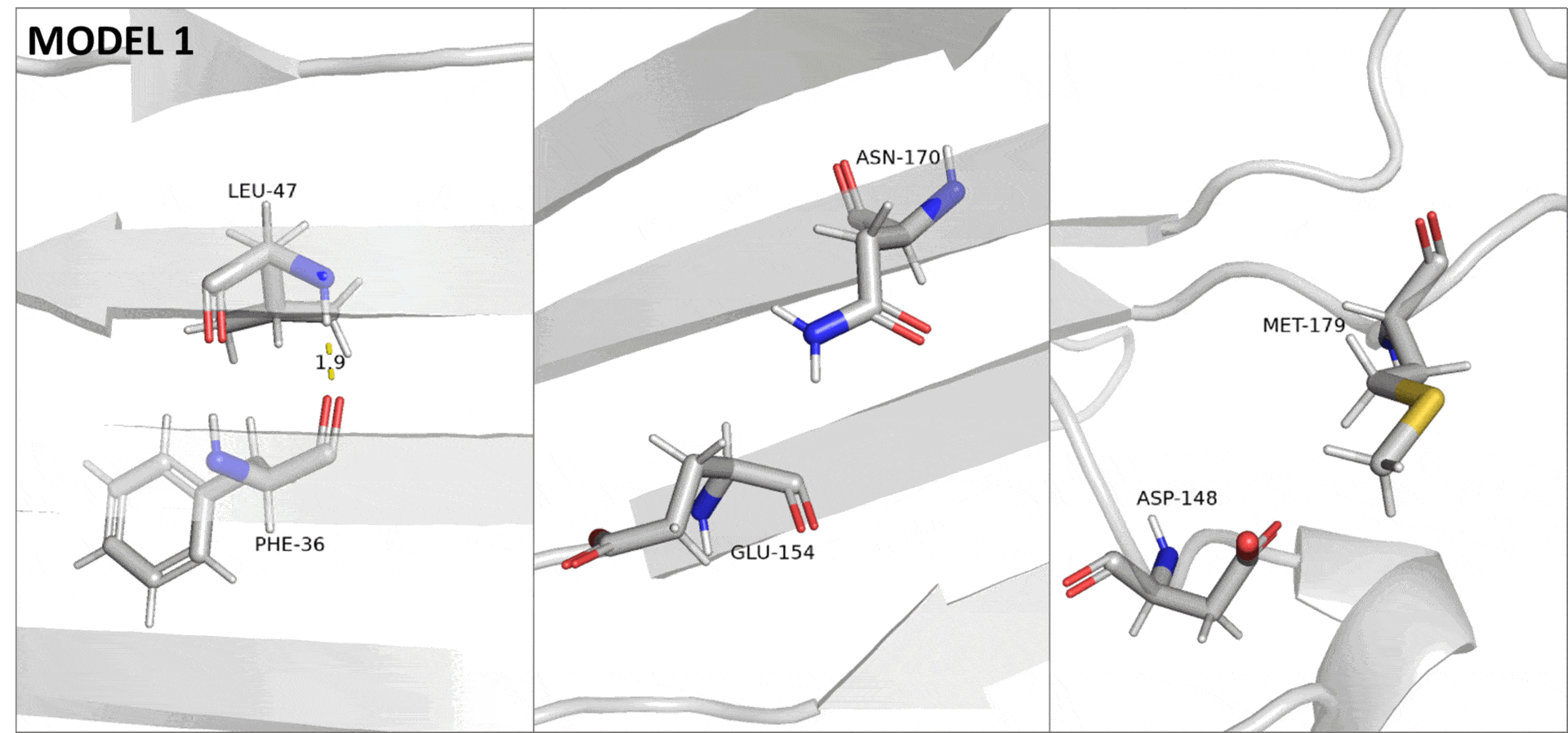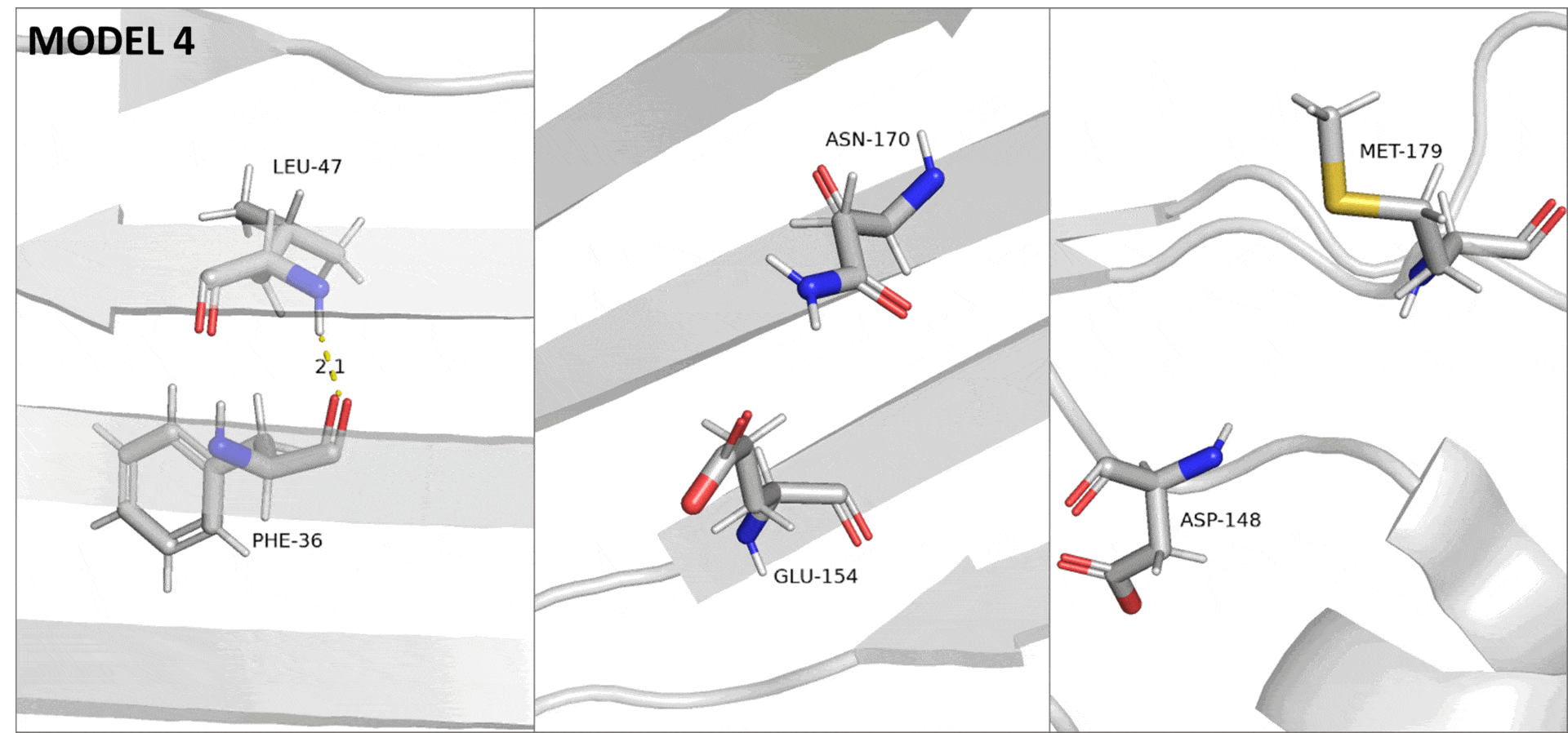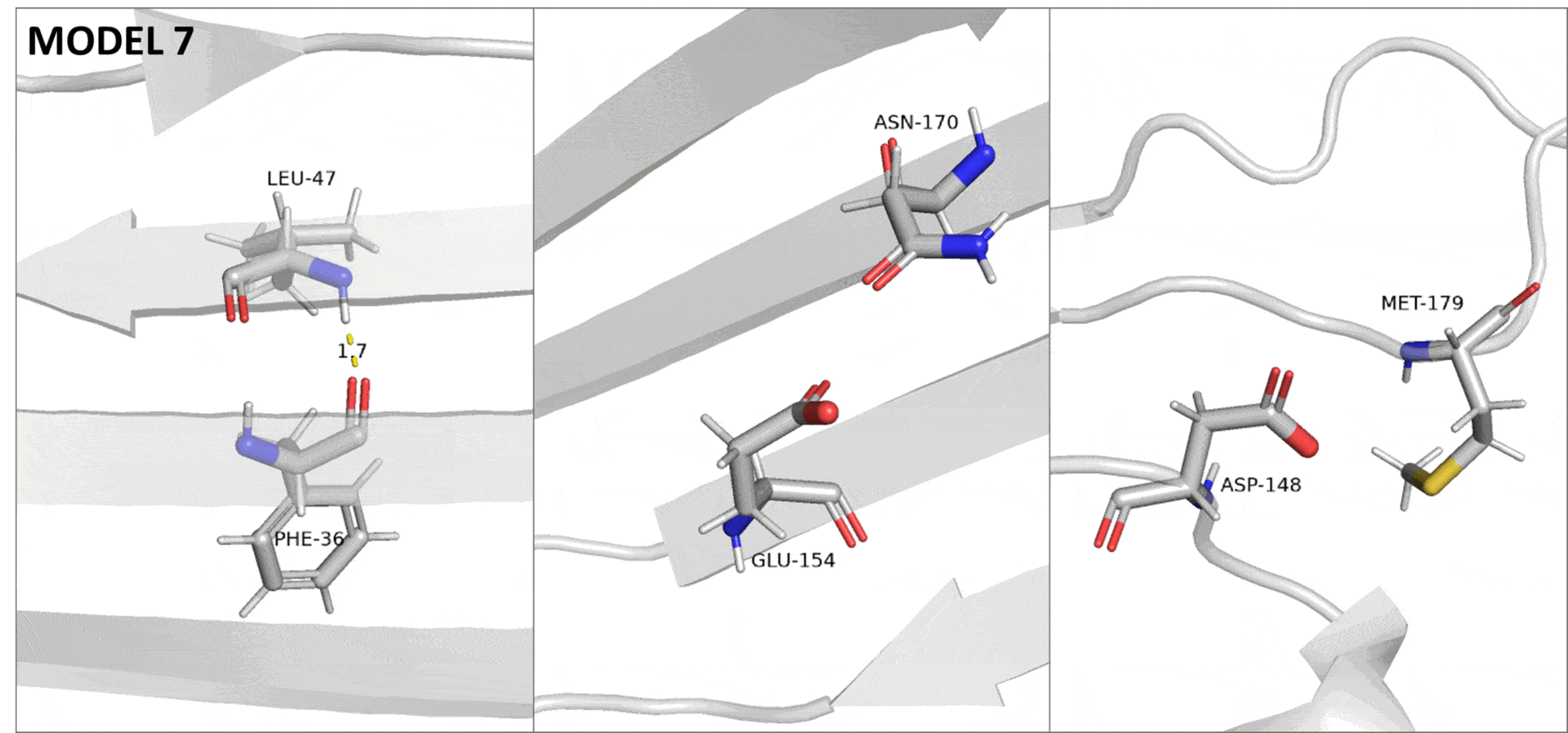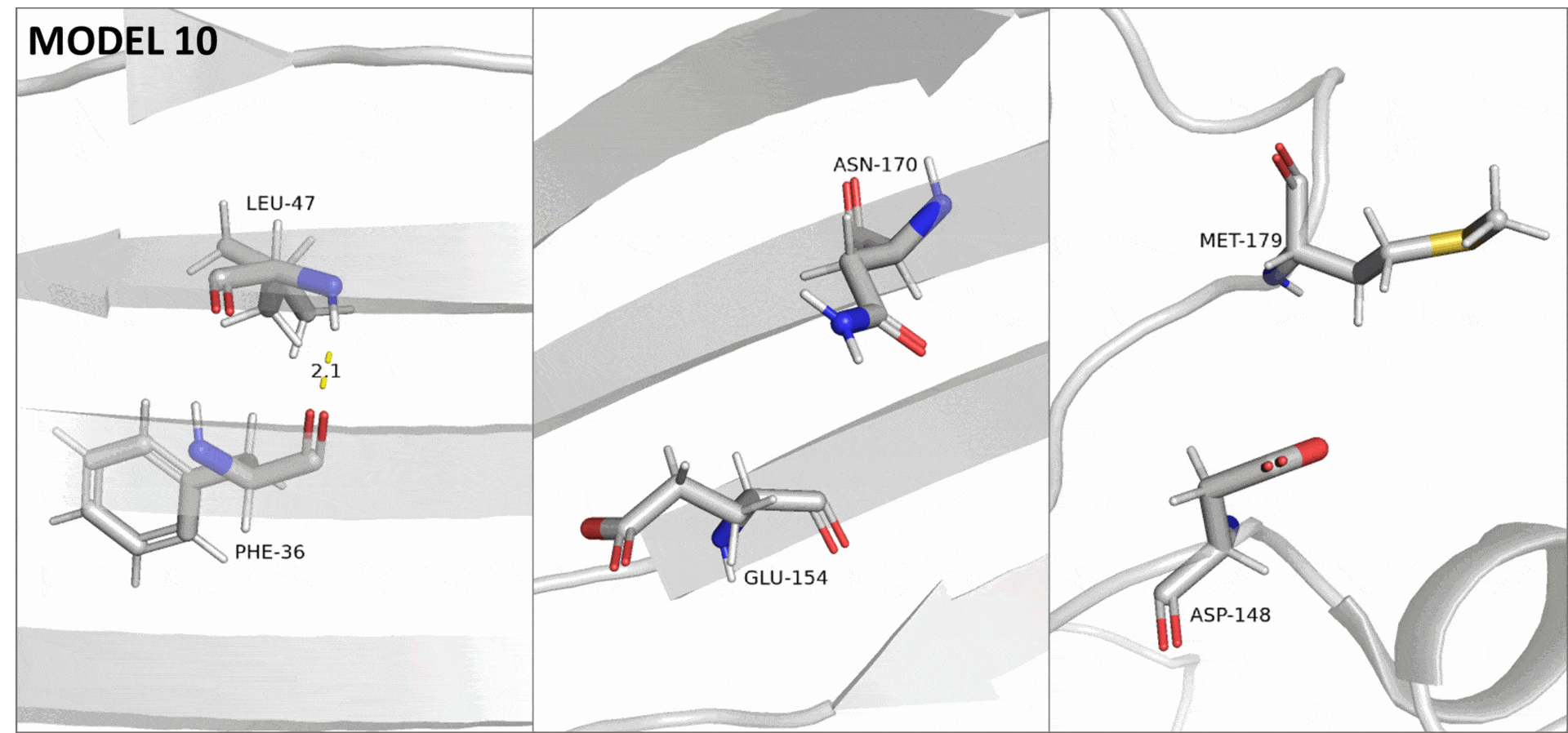
Для описания были выбраны три водородные связи в структуре, полученной методом РСА (Рис. 1). Из остава бета-бочонка была выбрана связь между азотом L47 и кислородом F36. В качестве взаимодействия между атомами боковых цепей элемента вторичной структуры была выбрана связь между азотом амидной группы N170 и кислородом карбоксильной группы E154. Оба этих остатка тоже входят в состав бета-тяжей. Также была взята связь между остатками неструктурированных петель: от азота M179 к кислороду боковой цепи D148. Так как белок представляет собой бета-бочонок, в растворе боковые цепи всех его остатков занимают поверхностное положение.
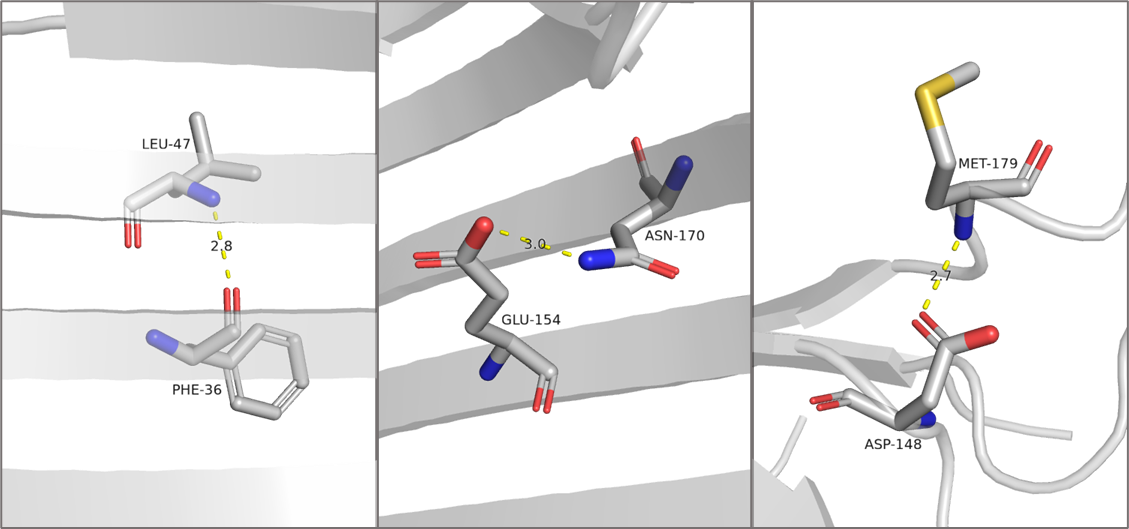
Для кристаллической структуры были измерены длины выбранных связей, в то время как для всех моделей ЯМР были получены донорно-акцепторные расстояния и посчитана статистика по встречаемости связей (Табл. 1).
| Параметр | Бета-лист, остав (L47 -> F36) |
Бета-лист, боковые цепи (N170 -> E154) |
Неструкт. петля (M179 -> D148) |
|---|---|---|---|
| Длина в кристалле (Å) | 2.84 | 3.01 | 2.70 |
| Число и % моделей ЯМР | 9 (90%) | 1 (10%) | 0 (0%) |
| Средняя длина (ЯМР) | 2.91 | 6.18 | 7.06 |
| Медианная длина (ЯМР) | 2.88 | 7.13 | 7.2 |
| Мин. длина (ЯМР) | 2.62 | 3.19 | 4.58 |
| Макс. длина (ЯМР) | 3.32 | 7.94 | 10.43 |
Из выбранных по кристаллической структуре связей только первые две, принадлежащие остаткам бета-бочонка, встречаются в структуре ЯМР. При этом связь между атомами остава имеется в 9 из 10 моделей, тогда как связь между боковыми цепями присутствует только в одной из них. Средние и медианные значения донорно-акцепторных расстояний для трех пар остатков в структуре ЯМР всегда оказываются больше длин соответствующих связей в кристалле. Все это объясняется тем, что в эксперименте по спектроскопии ЯМР исследуется структура белка в растворе, где подвижность его остатков гораздо выше, чем в кристалле.
Если сравнить распределения донорно-акцепторных расстояний в моделях ЯМР (Рис. 2) для выбранных связей, то можно заметить явный тренд. В случае остава бета-листа длины связей, а также их разброс, меньше всего. Далее по возрастанию идет пара остатков N170-E154 (бета-лист, боковые цепи), а после нее — пара M179-D148 (неструктурированные петли). Эта же закономерность прослеживается и по рассчитанным статистикам (Табл. 1): среднее, медианное, минимальное и максимальное значения для донорно-акцепторных расстояний (а также встречаемость водородных связей) возрастают от первой паре остатков к третьей.
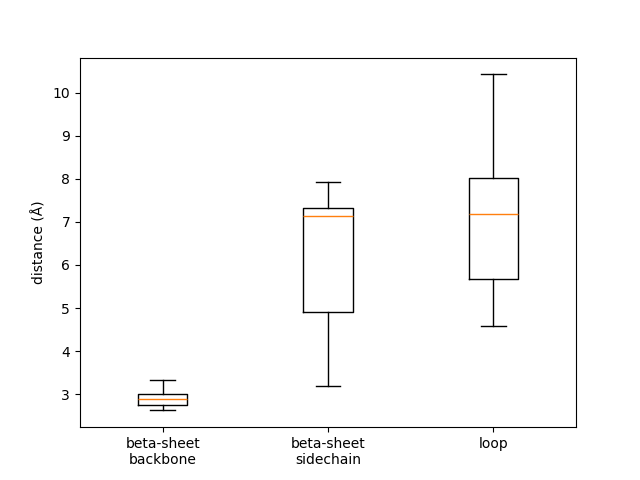
Эта разница объясняется различными уровнями подвижности разных участков белка. Остатки в составе структурированных участков белка часто занимают относительно устойчивое положение, так как элементы вторичной структуры поддерживаются многочисленными водородными связями между атомами остава и, соответственно, являются достаточно жесткими. При этом боковые цепи зачастую могут принимать различные конформации (особенно, если они смотрят в раствор), а атомы остава, непосредственно участвующие в поддержании структуры, имеют более-менее фиксированное положение. Изменение конформации остава даже одного остатка может прервать вторичную структуру, что повлечет большие потери в энергии. В то же время, неструктурированные петли могут быть достаточно подвижными, если не удерживаются достаточным количеством взаимодействий со стороны белка.
При получении структуры методом ЯМР неполнота данных, вызванная в том числе подвижностью белковой цепи, приводит к невозможности полностью разрешить структуру. На практике это проявляется в том, что для одного белка получается сразу много моделей, в первом приближении описывающих разные состояния белка в растворе. Поэтому в данном случае, различия в подвижности описываемых участков можно увидеть, переключаясь между разными моделями ЯМР (Рис. 3).
Сессия pymol для структуры 6OQH
Сессия pymol для структуры 2F1C
Команды pymol для структуры 6OQH
Команды pymol для структуры 2F1C
Скрипт для измерения расстояний в моделях ЯМР
Задание 2
Помимо подвижности цепи в растворе неполнота данных, возникающих при спектроскопии ЯМР, может быть вызвана и чисто техническими причинами: шумом и неоптимальными условиями проведения эксперимента. Для проверки того, насколько хорошо множество моделей ЯМР отражает реальную подвижность белка, было проведено сопоставление средних по остаткам значений RMSF, посчитанных по ансамблю моделей ЯМР, со средними значениями B-факторов, взятых из структуры, полученной методом РСА.
Для начала было произведено сравнение последовательностей, соответствующих двум структурам. Для этого было получено выравнивание двух структур в pymol, а также списки остатков, для атомов которых есть координаты в cif-файлах. После их сопоставления оказалось, что последовательности не полностью соответствуют друг другу. В белке, использованном в рентгеновском эксперименте (2F1C) отсутствует N-концевой участок из 3 остатков и имеется гексагистидиновый тэг на C-конце. По отношению к структуре 6OQH в 2F1C также не определены координаты атомов остатков в четырех отдельных участках внутри последовательности. В то же время, в белке, для которого была получена ЯМР структура, имеется делеция в области петли 6 (она была введена специально для изучения ее влияния на подвижность остатков).
После выравнивания остатков и проверки правильности их сопоставления были рассчитаны средние значения квадратов RMSF и B-факторов для всех остатков. Зависимость между ними представлена на рисунке ниже (Рис. 4).
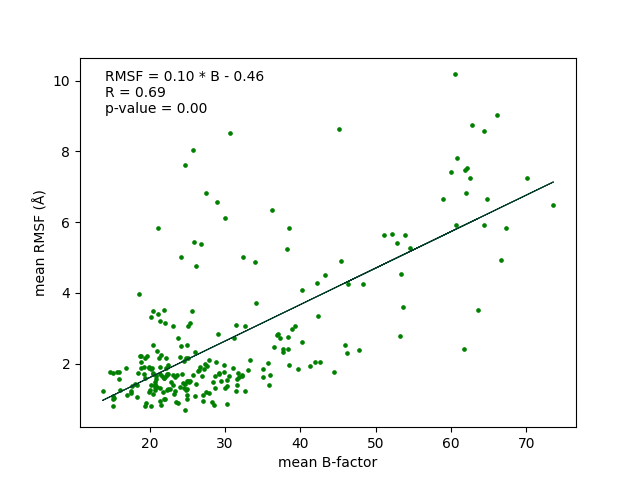
Можно заметить, что между измеренными параметрами линейная зависимость с неплохой корреляцией (0.65) и низким значением p-value для наклона прямой. Для реалистичного ансамбля моделей ЯМР зависимость является линейной (относительно квадрата RMSF) и описывается формулой
RMSF2 = 3*B / (8 * ℼ²)
Это свидетельствует о том, что в данном случае ансамбль неплохо отражает подвижность остатков белка.