Молекулярная динамика биологических молекул в GROMACS¶
import os
os.chdir('/home/students/y15/smargasyuk')
Построим решетку из 64 (4 4 4) молекул липида (заменим 34 на 64 в файле b.top), сдвинем и оптимизируем структуру
Выдача последней команды:
:-) G R O M A C S (-:
GROningen Mixture of Alchemy and Childrens' Stories
:-) VERSION 4.5.5 (-:
Written by Emile Apol, Rossen Apostolov, Herman J.C. Berendsen,
Aldert van Buuren, Pär Bjelkmar, Rudi van Drunen, Anton Feenstra,
Gerrit Groenhof, Peter Kasson, Per Larsson, Pieter Meulenhoff,
Teemu Murtola, Szilard Pall, Sander Pronk, Roland Schulz,
Michael Shirts, Alfons Sijbers, Peter Tieleman,
Berk Hess, David van der Spoel, and Erik Lindahl.
Copyright (c) 1991-2000, University of Groningen, The Netherlands.
Copyright (c) 2001-2010, The GROMACS development team at
Uppsala University & The Royal Institute of Technology, Sweden.
check out http://www.gromacs.org for more information.
This program is free software; you can redistribute it and/or
modify it under the terms of the GNU General Public License
as published by the Free Software Foundation; either version 2
of the License, or (at your option) any later version.
:-) mdrun (-:
Option Filename Type Description
------------------------------------------------------------
-s b_em.tpr Input Run input file: tpr tpb tpa
-o b_em.trr Output Full precision trajectory: trr trj cpt
-x b_em.xtc Output, Opt. Compressed trajectory (portable xdr format)
-cpi b_em.cpt Input, Opt. Checkpoint file
-cpo b_em.cpt Output, Opt. Checkpoint file
-c b_em.gro Output Structure file: gro g96 pdb etc.
-e b_em.edr Output Energy file
-g b_em.log Output Log file
-dhdl b_em.xvg Output, Opt. xvgr/xmgr file
-field b_em.xvg Output, Opt. xvgr/xmgr file
-table b_em.xvg Input, Opt. xvgr/xmgr file
-tablep b_em.xvg Input, Opt. xvgr/xmgr file
-tableb b_em.xvg Input, Opt. xvgr/xmgr file
-rerun b_em.xtc Input, Opt. Trajectory: xtc trr trj gro g96 pdb cpt
-tpi b_em.xvg Output, Opt. xvgr/xmgr file
-tpid b_em.xvg Output, Opt. xvgr/xmgr file
-ei b_em.edi Input, Opt. ED sampling input
-eo b_em.edo Output, Opt. ED sampling output
-j b_em.gct Input, Opt. General coupling stuff
-jo b_em.gct Output, Opt. General coupling stuff
-ffout b_em.xvg Output, Opt. xvgr/xmgr file
-devout b_em.xvg Output, Opt. xvgr/xmgr file
-runav b_em.xvg Output, Opt. xvgr/xmgr file
-px b_em.xvg Output, Opt. xvgr/xmgr file
-pf b_em.xvg Output, Opt. xvgr/xmgr file
-mtx b_em.mtx Output, Opt. Hessian matrix
-dn b_em.ndx Output, Opt. Index file
-multidir b_em Input, Opt., Mult. Run directory
Option Type Value Description
------------------------------------------------------
-[no]h bool no Print help info and quit
-[no]version bool no Print version info and quit
-nice int 0 Set the nicelevel
-deffnm string b_em Set the default filename for all file options
-xvg enum xmgrace xvg plot formatting: xmgrace, xmgr or none
-[no]pd bool no Use particle decompostion
-dd vector 0 0 0 Domain decomposition grid, 0 is optimize
-nt int 0 Number of threads to start (0 is guess)
-npme int -1 Number of separate nodes to be used for PME, -1
is guess
-ddorder enum interleave DD node order: interleave, pp_pme or cartesian
-[no]ddcheck bool yes Check for all bonded interactions with DD
-rdd real 0 The maximum distance for bonded interactions with
DD (nm), 0 is determine from initial coordinates
-rcon real 0 Maximum distance for P-LINCS (nm), 0 is estimate
-dlb enum auto Dynamic load balancing (with DD): auto, no or yes
-dds real 0.8 Minimum allowed dlb scaling of the DD cell size
-gcom int -1 Global communication frequency
-[no]v bool yes Be loud and noisy
-[no]compact bool yes Write a compact log file
-[no]seppot bool no Write separate V and dVdl terms for each
interaction type and node to the log file(s)
-pforce real -1 Print all forces larger than this (kJ/mol nm)
-[no]reprod bool no Try to avoid optimizations that affect binary
reproducibility
-cpt real 15 Checkpoint interval (minutes)
-[no]cpnum bool no Keep and number checkpoint files
-[no]append bool yes Append to previous output files when continuing
from checkpoint instead of adding the simulation
part number to all file names
-maxh real -1 Terminate after 0.99 times this time (hours)
-multi int 0 Do multiple simulations in parallel
-replex int 0 Attempt replica exchange periodically with this
period (steps)
-reseed int -1 Seed for replica exchange, -1 is generate a seed
-[no]ionize bool no Do a simulation including the effect of an X-Ray
bombardment on your system
Getting Loaded...
Reading file b_em.tpr, VERSION 4.5.5 (single precision)
Starting 8 threads
Loaded with Money
Making 3D domain decomposition 2 x 2 x 2
Steepest Descents:
Tolerance (Fmax) = 1.00000e+00
Number of steps = 1000000
Step= 0, Dmax= 2.0e-02 nm, Epot= 4.74007e+05 Fmax= 4.37970e+05, atom= 1842
Step= 1, Dmax= 2.0e-02 nm, Epot= 7.19293e+04 Fmax= 3.57933e+04, atom= 2588
Step= 2, Dmax= 2.4e-02 nm, Epot= 3.56795e+04 Fmax= 9.52568e+03, atom= 2993
Step= 3, Dmax= 2.9e-02 nm, Epot= 3.46459e+04 Fmax= 1.50508e+04, atom= 2992
Step= 4, Dmax= 3.5e-02 nm, Epot= 2.99879e+04 Fmax= 1.37059e+04, atom= 2592
Step= 6, Dmax= 2.1e-02 nm, Epot= 1.30782e+04 Fmax= 4.02791e+03, atom= 1342
Step= 9, Dmax= 6.2e-03 nm, Epot= 1.04672e+04 Fmax= 1.87751e+03, atom= 2392
Step= 10, Dmax= 7.5e-03 nm, Epot= 1.02199e+04 Fmax= 3.76147e+03, atom= 1792
Step= 11, Dmax= 9.0e-03 nm, Epot= 1.01147e+04 Fmax= 4.34365e+03, atom= 2392
Step= 13, Dmax= 5.4e-03 nm, Epot= 7.19423e+03 Fmax= 1.91912e+03, atom= 2765
Step= 15, Dmax= 3.2e-03 nm, Epot= 6.81202e+03 Fmax= 2.03533e+03, atom= 2765
Step= 16, Dmax= 3.9e-03 nm, Epot= 6.37647e+03 Fmax= 3.00818e+03, atom= 2765
Step= 17, Dmax= 4.6e-03 nm, Epot= 6.07244e+03 Fmax= 2.75324e+03, atom= 2765
Step= 19, Dmax= 2.8e-03 nm, Epot= 5.60144e+03 Fmax= 8.43337e+02, atom= 2765
Step= 20, Dmax= 3.3e-03 nm, Epot= 5.49136e+03 Fmax= 3.08327e+03, atom= 2765
Step= 21, Dmax= 4.0e-03 nm, Epot= 5.09456e+03 Fmax= 2.09083e+03, atom= 2765
Step= 23, Dmax= 2.4e-03 nm, Epot= 4.83141e+03 Fmax= 8.78173e+02, atom= 2765
Step= 25, Dmax= 1.4e-03 nm, Epot= 4.78063e+03 Fmax= 1.07042e+03, atom= 2765
Step= 26, Dmax= 1.7e-03 nm, Epot= 4.51750e+03 Fmax= 1.00741e+03, atom= 2765
Step= 27, Dmax= 2.1e-03 nm, Epot= 4.46707e+03 Fmax= 1.77657e+03, atom= 2765
Step= 28, Dmax= 2.5e-03 nm, Epot= 4.35788e+03 Fmax= 1.28547e+03, atom= 915
Step= 30, Dmax= 1.5e-03 nm, Epot= 4.20725e+03 Fmax= 6.94208e+02, atom= 915
Step= 31, Dmax= 1.8e-03 nm, Epot= 3.97280e+03 Fmax= 1.39504e+03, atom= 915
Step= 32, Dmax= 2.2e-03 nm, Epot= 3.97165e+03 Fmax= 1.44422e+03, atom= 915
Step= 33, Dmax= 2.6e-03 nm, Epot= 3.85030e+03 Fmax= 1.70904e+03, atom= 915
Step= 35, Dmax= 1.6e-03 nm, Epot= 3.71519e+03 Fmax= 3.29197e+02, atom= 472
Step= 36, Dmax= 1.9e-03 nm, Epot= 3.32876e+03 Fmax= 1.57250e+03, atom= 915
Step= 39, Dmax= 5.6e-04 nm, Epot= 3.23180e+03 Fmax= 8.45033e+02, atom= 2765
Step= 42, Dmax= 1.7e-04 nm, Epot= 3.20617e+03 Fmax= 6.21543e+02, atom= 9153
Step= 50, Dmax= 1.6e-06 nm, Epot= 3.20591e+03 Fmax= 6.19379e+02, atom= 9155
Step= 51, Dmax= 1.9e-06 nm, Epot= 3.20911e+03 Fmax= 6.16887e+02, atom= 2765
Stepsize too small, or no change in energy.
Converged to machine precision,
but not to the requested precision Fmax < 1
Double precision normally gives you higher accuracy.
writing lowest energy coordinates.
Steepest Descents converged to machine precision in 52 steps,
but did not reach the requested Fmax < 1.
Potential Energy = 3.2059077e+03
Maximum force = 6.1937860e+02 on atom 915
Norm of force = 1.7839124e+02
gcq#246: "Drugs are Bad, mmokay" (South Park)Максимальная сила изменилась от 3.37e+05 до 6.17e+02: уменьшилась в ~1000 раз.
Добавим молекулы воды типа spc и оптимизируем структуру:
genbox -cp b_em -p b -cs spc216 -o b_s
grompp -f pr -c b_s -p b -o b_pr -maxwarn 1
mdrun -deffnm b_pr -v
Опишем изменения структуры между файлами b_s и b_pr. Произошла оптимизация конформации молекул липида: в файле b_s они все имеют одинаковую конформацию (т.е. являются сдвинутыми копиями одной молекулы), в b_pr конформация молекул отличается.
Запустим задачу:
grompp -f md -c b_pr -p b -o b_md -maxwarn 1
sbatch -N1 --ntasks-per-node=2 -e error-gpu.log -o output.log -t 350 -p gpu impi /opt/ccoe/gromacs-5.0.4/build/bin/gmx_mpi mdrun -testverlet -deffnm b_md -v
Анализ результатов¶
Ссылка на лог GROMACS: md.log.
Опишем параметры системы:
- Силовое поле: см. описание ниже
- Заряд системы: 0
- Ячейка -- параллелепипед, размер 6.26 4.443 5.778
Оптимизация энергии¶
!cat smargasyuk/em.mdp
Параметры:
- метод оптимизации энергии: steep;
- расчет электростатики: Кулон;
- расчет Ван-дер-Ваальсовых сил: Леннард-Джонс;
Растворитель¶
!cat smargasyuk/pr.mdp
Параметры:
- модель растворителя: spc (трехточечная модель);
- 1000 шагов утряски растворителя;
- длина шага 0.0002 пикосекунды;
- термостат: velocity rescale;
- баростат: нет;
- электростатика: pme;
- Ван-дер-Ваальсовы силы: Леннард-Джонс
Основное моделирование¶
!cat smargasyuk/smargasyuk/md.mdp
- Время моделирования: 6 часов 13 минут;
- Число процессоров: 16 (2 MPI ranks * 8 MPI threads);
- Эффективность масштабирования: 98.9% (Part of the total run time spent waiting due to load imbalance: 1.1 %);
- Число шагов: 10 ** 7;
- Длина шага: 0.005 пс;
- Алгоритм интегратора: md;
- Расчет электростатики и ВдВ сил: pme и Леннард-Джонс;
- Термостат и баростат: velocity rescale и Берендсен
Результат моделирования¶
Визуализируем движение молекул:
trjconv -f b_md.xtc -s b_md.tpr -o b_pbc_1.pdb -skip 20 -pbc mol
Сборка бислоя завершается примерно к 22 кадру (время 10500 пикосекунд).
Построим зависимость размера ячейки от времени:
g_traj -f b_md.xtc -s b_md.tpr -ob box_1.xvg
Ось Z -- нормаль к поверхности бислоя, построим зависимость площади между двумя другими осями от времени
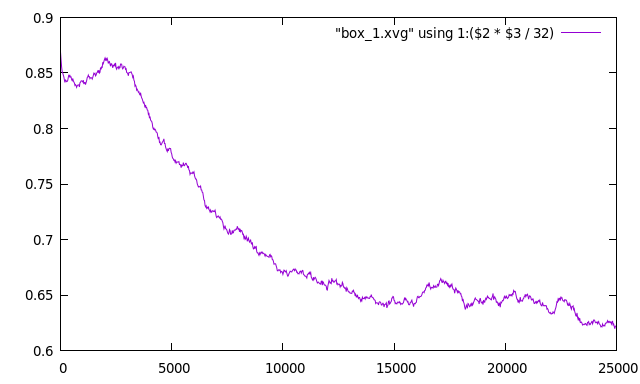
При сборке слоя площадь , занимаемая молекулой (произведение размеров по другим осям) понижается с 0.85 до 0.625.
Построим график изменения площади гидрофильной и гидрофобной поверхности:
g_sas -f b_md.xtc -s b_md.tpr -o sas_b.xvg
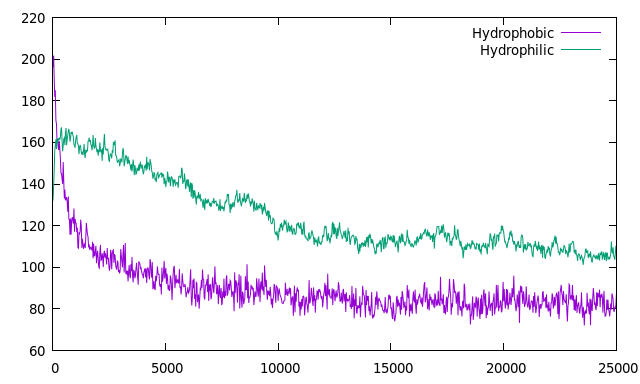
Сборка бислоя сопровождается резким (в начале) падением площади гидрофобной поверхности, гидрофильная поверхность снижается слабее.
Построим значения меры порядка в начале и конце симуляции:
g_order -s b_md -f b_md.xtc -o ord_start.xvg -n sn1.ndx -e 5000 -d z
g_order -s b_md -f b_md.xtc -o ord_end.xvg -n sn1.ndx -b 20000 -d z
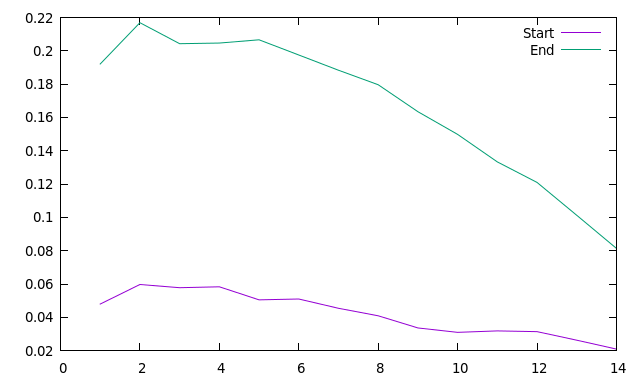
На обоих графиках упорядоченность падает с увеличением номера атома, в конце симуляции это происходит быстрее, т.к. после сборки бислоя гидрофильные головки молекул оказываются закреплены.