Практикум 3¶
Импортировали rdkit и функции IPython для отображения картинок в ноутбуке.
from rdkit import Chem
from rdkit.Chem import AllChem
from rdkit import RDConfig
from rdkit.Chem.Draw import IPythonConsole
from rdkit.Chem import Draw
from IPython.display import display,Image
Создали объект rdkit из SMILES-формулы ибупрофена, изобразили структурную формулу.
ibu=Chem.MolFromSmiles('CC(C)CC1=CC=C(C=C1)C(C)C(=O)O')
AllChem.Compute2DCoords(ibu)
display(ibu)

Добавили терминальную тройную связь к молекуле ибупрофена: так она может вступать в реакцию азид-алкинового циклоприсоединения с азидами.
ibu_alkine=Chem.MolFromSmiles('CC(C)CC1=CC=C(C=C1)C(CC#C)C(=O)O')
AllChem.Compute2DCoords(ibu_alkine)
display(ibu_alkine)

Построили шаблон для симуляции реакции циклоприсоединения: структурную формулу одного из продуктов реакции циклоприсоединения азида с молекулой, изображенной выше, можно получить заменой азидной группы на этот шаблон.
template=Chem.MolFromSmiles('CC(C)CC1=CC=C(C=C1)C(CC1=CN=NN1)C(=O)O')
AllChem.Compute2DCoords(template)
display(template)

Изменили нумерацию в формуле SMILES шаблона: теперь формулу SMILES продукта реакции можно получить заменой азидной группы в SMILES формуле азида на SMILES формулу шаблона.
template=Chem.MolFromSmiles('N1N=NC=C(CC(C(=O)O)C2=CC=C(CC(C)C)C=C2)1')
AllChem.Compute2DCoords(template)
display(template)
template1 = '(N1N=NC=C(CC(C(=O)O)C2=CC=C(CC(C)C)C=C2)1)'

Рассчитали параметры правила четырёх Липински для ибупрофена.
import rdkit.Chem.Lipinski as Lipinksy
print(Lipinksy.NumHDonors(ibu))
print(Lipinksy.NumHAcceptors(ibu))
print(Lipinksy.rdMolDescriptors.CalcExactMolWt(ibu))
print(Lipinksy.rdMolDescriptors.CalcCrippenDescriptors(ibu)[0])
Загрузили из PubChem все соединения с азидной группой (запрос по SMILES 'N=[N+]=[N-]', при поиске учитывали заряд).
import os
os.system('wget {0}'.format('ftp://ftp-private.ncbi.nlm.nih.gov/pubchem/.fetch/30/471641666924966801.txt'))
Загрузили полученный файл в массив numpy, затем выбрали только формулы длины не более 30, содержащие 1 молекулу.
import numpy as np
strings=np.genfromtxt('pr3/41666924966801.txt',dtype=np.str)
smiles = []
for line in strings:
if len(line[1]) < 30 and not '.' in line[1]:
smiles.append(line[1])
len(smiles)
Создали функцию для проверки правила пяти Липински: по сравнению с правилом четырёх наложено дополнительное ограничение на количество связей, вокруг которых возможно вращение.
def lipinski_test(mol):
if (Lipinksy.NumHDonors(mol) < 5) & \
(Lipinksy.NumHAcceptors(mol) < 10) & \
(Lipinksy.rdMolDescriptors.CalcExactMolWt(mol) < 500) &\
(Lipinksy.rdMolDescriptors.CalcCrippenDescriptors(ibu)[0] < 5) & \
(Lipinksy.NumRotatableBonds(mol) < 10):
return True
else:
return False
Для всех формул азидов заменили азидную группу на шаблон. Не все полученные формулы оказались корректными (например, в формуле азида мог содержаться цикл, и нумерация в цикле вступала в конфликт с нумерацией в шаблоне). Получилось 5432 симулированных продукта реакции, удовлетворяющих правилу пяти Липински, для 14312 формул азидов, поданных на вход.
smiles_repl = []
for smi in smiles[:15000]:
if 'N=[N+]=[N-]' in smi:
newsmi=smi.replace('N=[N+]=[N-]',template1)
else:
continue
# Новую молекулу лучше создавать в try из-за битых Smiles
try:
newmol=Chem.MolFromSmiles(newsmi)
if lipinski_test(newmol) == True:
smiles_repl.append(newmol)
except:
pass
len(smiles_repl)
Отобразили некоторые полученные продукты.
from IPython.display import SVG
for m in smiles_repl:
AllChem.Compute2DCoords(m)
pil = Draw.MolsToGridImage(smiles_repl, useSVG=True, molsPerRow=3, maxMols=12, subImgSize=(500, 500))
display(pil)
Построили карту сходства ибупрофена с одним из полученных продуктов.
from rdkit.Chem.Draw import SimilarityMaps
fp = SimilarityMaps.GetMorganFingerprint(ibu, fpType='bv')
fig, maxweight = SimilarityMaps.GetSimilarityMapForFingerprint(ibu, smiles_repl[4], SimilarityMaps.GetMorganFingerprint)

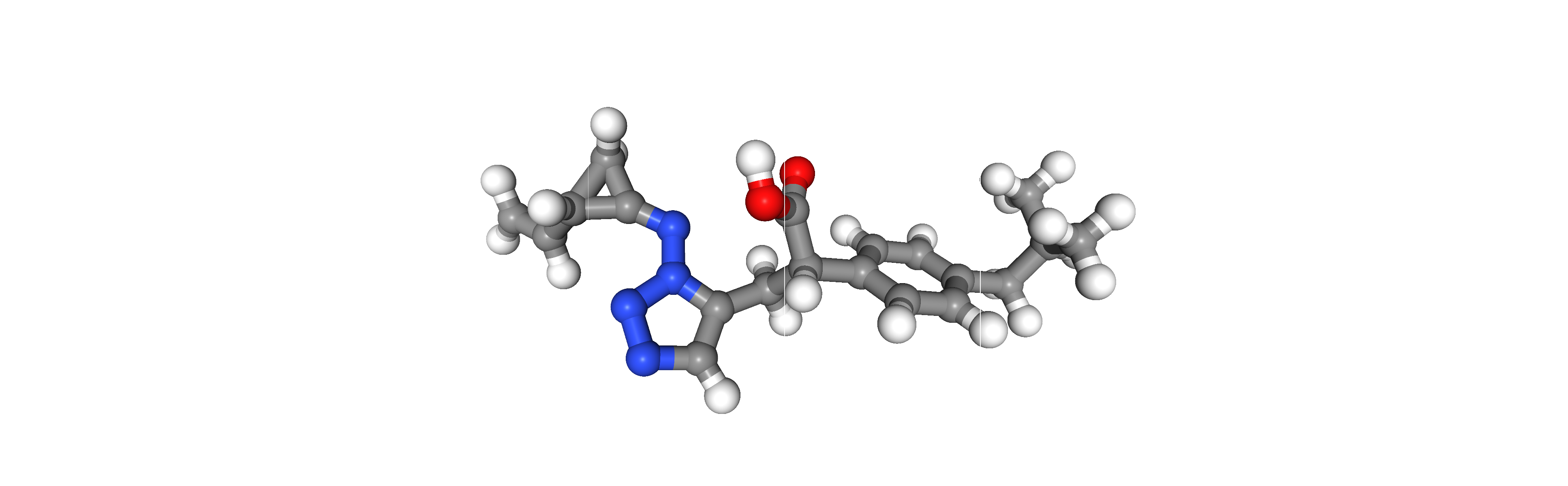
Построили трёхмерную структуру полученной молекулы
m = smiles_repl[4]
m3d=Chem.AddHs(m)
Chem.AllChem.EmbedMolecule(m3d)
AllChem.MMFFOptimizeMolecule(m3d,maxIters=500,nonBondedThresh=200 )
m3d

Показали эту структуру в виджете nglview
import nglview as nv
import ipywidgets
w = nv.show_rdkit(m3d)
w
w.download_image()
Виджет не отображается в HTML, трехмерная структура выглядит так: