Для выполнения данного задания был использован набор чтений 16-й хромосомы человека. Для него были выполнены анализ и очистка чтений, проведено картирование очищенного набора чтений на хромосому из сборки генома человека hg19, найдены и аннотированы отличия от референсного генома.
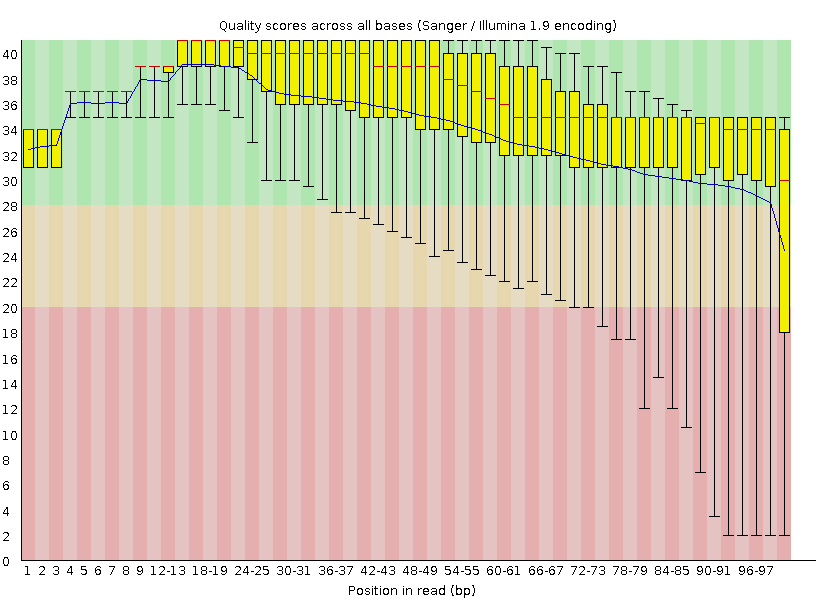
Анализ исходного набора чтений был проведен с помощью программы FastQC. Диаграмма 1, полученная в результате работы этой программы, отражает качество нуклеотидов в чтениях набора.
Согласно диаграмме, большинство прочтенных нуклеотидов имеет приемлемое качество (нижний квартиль почти всюду выше красной зоны): таким образом, эксперимент корректен. Однако в концевых позициях многих ридов качество нуклеотидов ниже допустимого, поэтому требуется очистка чтений.
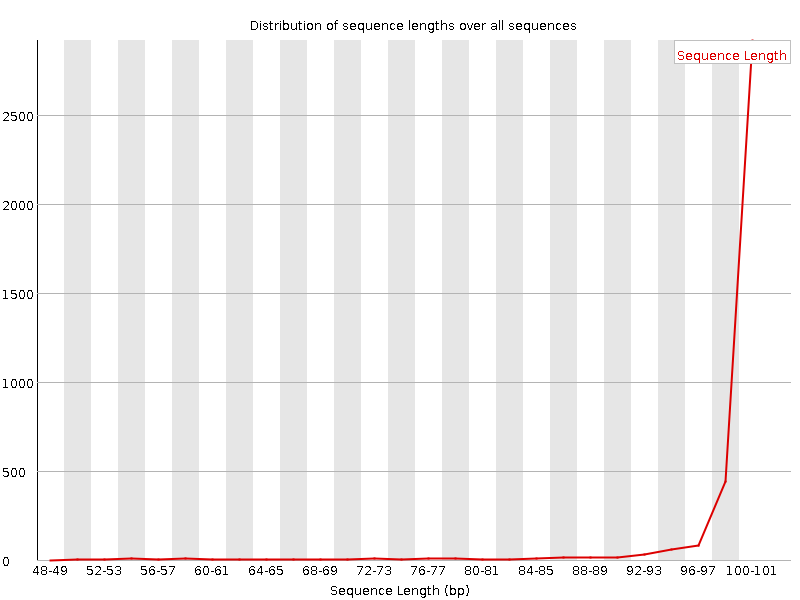
Для очистки были удалены концевые позиции чтений с качеством ниже 20; после этого чтения, длина которых ниже 50, были удалены (это позволит избежать неоднозначного картирования). Для этого была применена программа Trimmomatic с методом очистки TRAILING:20 MINLEN:50. После очистки из 3965 чтений исходного набора было сохранены 3798 (96%). Диаграмма 2 описывает качество чтений после очистки.
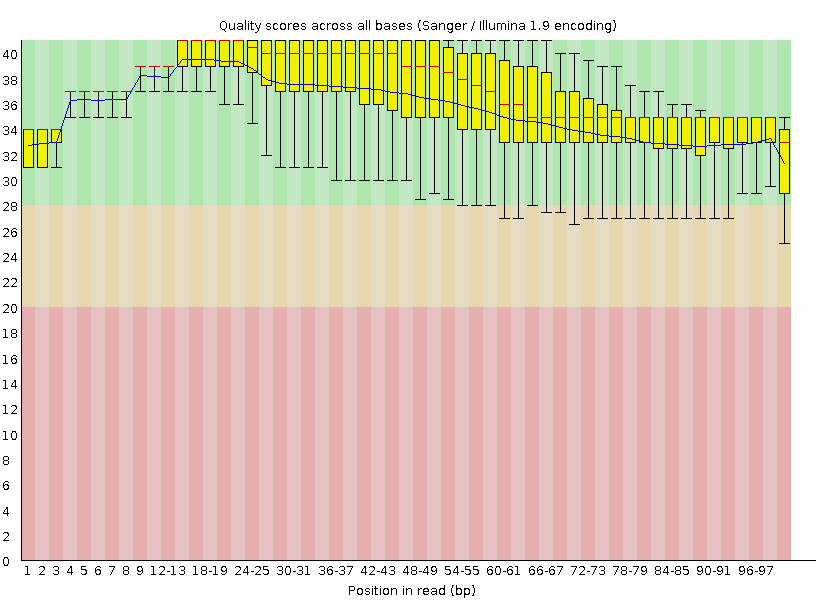
Как и ожидалось, после очистки значительно улучшились показатели качества чтений концевых нуклеотидов: теперь качество всех нуклеотидов находится в пределах допустимого (Phred > 20).
Сравним другие показатели исходного и очищенного наборов. На графике распределения последовательностей по среднему качеству нуклеотида при очистке удалены все последовательности со средним качеством ниже 20 (обычно качество концевых нуклеотидов ниже всего: последовательности с низким средним качеством имеют низкое качество концевых нуклеотидов) и незначительное количество прочих (вид правой части графика практически не меняется).
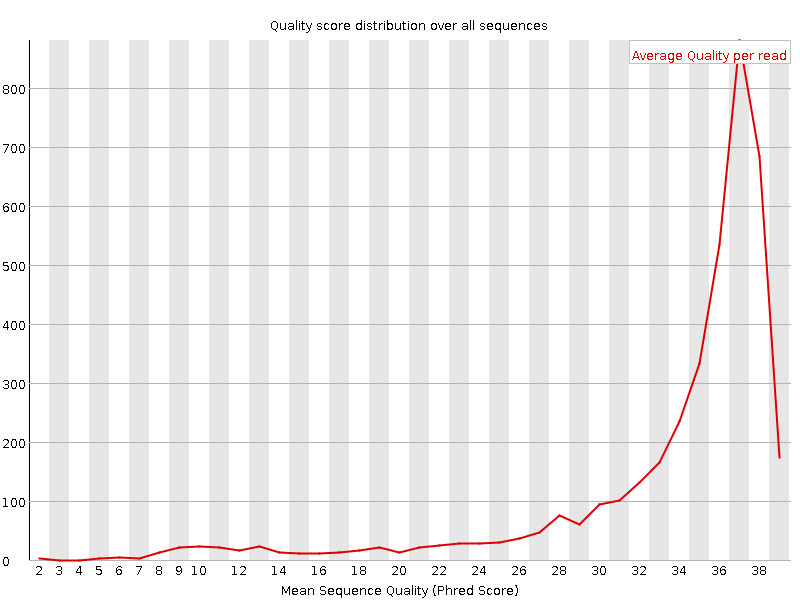
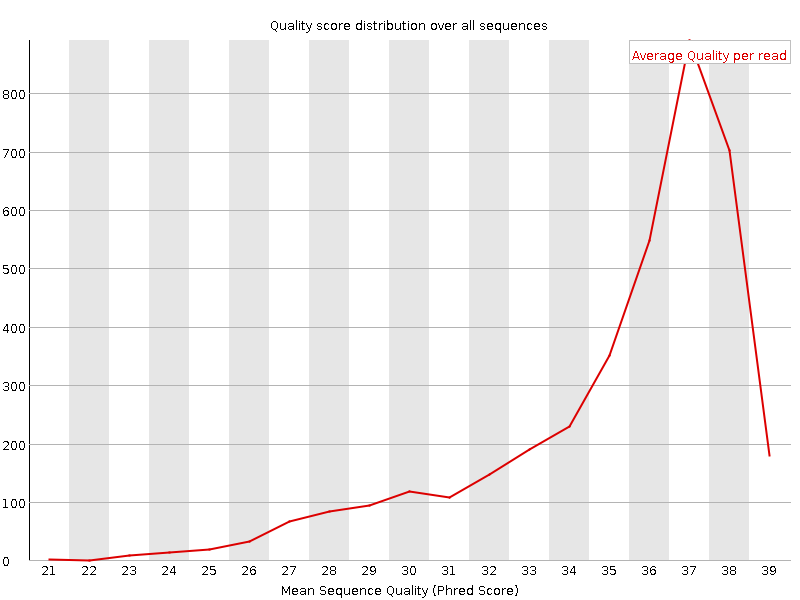
Кроме того, вследствие метода очистки изменились длины последовательностей: все последовательности исходного набора имеют длину 100, в то время как в очищенном наборе длина принимает значения от 50 до 100 (при этом большинство чтений имеет длину 100).
Картирование очищенного набора чтений с помощью программы BWA производилось на референсную последовательность 16-й хромосомы человека из hg19. Для построения и анализа выравнивания были выполнены следующие шаги:
По данным программы (второе число в выдаче idxstats) все 3798 ридов были картированы на хромосому. Найдем среднее покрытие для одного из экзонов. Нуклеотид 56970672 имеет покрытие 101, этот показатель значительно выше среднего (данные получены из выдачи команды samtools depth align-sorted.bam >nucl-stat.tsv). Диаграмма 3 описывает распределение нуклеотидов по покрытию.
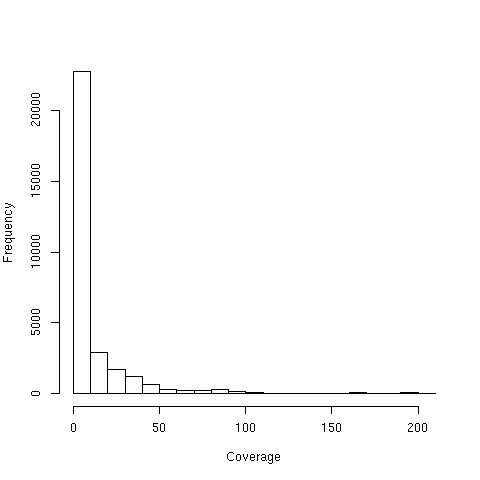
Нуклеотид принадлежит четвертому экзону гена HERPUD1 (экзон имеет координаты в геноме 56970599..56970733, направлен положительно). Среднее покрытие этого экзона равно 89. (данные получены из выдачи команды samtools depth -r chr16:56970599-56970733 align-sorted.bam > exon-stat.tsv)
Для поиска и визулизации различий между референсным геномом и чтениями были применены следующие команды:
Всего было найдено 63 SNP (однонуклеотидных полиморфизмов) и 2 инделя. Характеристики некоторых из найденных отличий приведены в таблице 1.
| № | координаты | тип полиморфизма | референсная последовательность | прочтенная последовательность | глубина покрытия | качество чтений |
| 1 | 11348246:11348251 | вставка | ACACTC | ACACTCACTC | 5 | 60 |
| 2 | 11367143 | замена | G | A | 36 | 222 |
| 3 | 56969148 | замена | G | A | 80 | 225 |
Для аннотации найденных полиморфизмов использовалась программа ANNOVAR. Далее перечислены команды, с помощью которых был выполнен запуск:
Опишем полученные результаты. База данных RefGene делит SNP по их расположению относительно генов: 5' или 3' нетранслируемые области (3 и 3 полиморфизма соответственно), 5' и 3' фланкирующие области (5 и 2 полиморфизма), экзоны (9), интроны (23) и межгенные области (18). SNP присутствуют в экзонах генов PRM3, PRM2, PRM1 (гены протаминов — белков, заменяющих гистоны в ходе сперматогенеза), TNP2 (переходный белок при замене гистонов на протамины), RMI2 (часть комплекса восстановления ДНК через гомологичную рекомбинацию), PRSS53 (протеаза, разделяющая белки по серину 53), HERPUD1 (ген, участвующий в ответе на ЭПР-стресс); SNP присутствуют в других участках тех же генов и SOCS1. Кроме того, выдача аннотирования по RefGene содержит данные о заменах аминокислот, соответствующих SNP в экзонах. В таблице 2 приведено описание несинонимичных SNP.
| № | ген | референсная последовательность | прочтенная последовательность | координата нуклеотида | референсная аминокислота | прочтенная аминокислота | координата аминокислоты |
| 1 | TNP2 | C | T | 391 | R | W | 131 |
| 2 | PRM3 | C | T | 310 | R | X (стоп-кодон) | 104 |
| 3 | PRM3 | G | A | 299 | R | Q | 100 |
| 4 | PRM2 | C | T | 290 | P | L | 97 |
| 5 | PRM2 | C | T | 281 | A | V | 94 |
| 6 | PRSS53 | C | G | 1216 | P | A | 406 |
| 7 | HERPUD1 | G | A | 149 | R | H | 50 |
Аннотация SNP по базе данных dbsnp содержит данные о вхождении мутаций в rs — кластер, содержащий все сообщения о данной мутации (мутации принадлежат rs, если о них когда-либо сообщали в dbsnp). Из 63 SNP в rs содержатся 57.
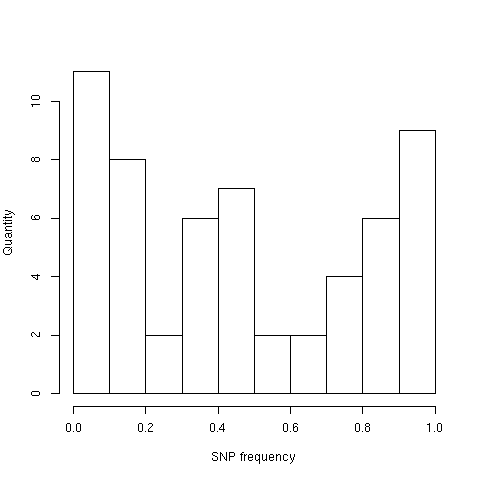
Аннотация SNP по 1000genomes содержит данные о частоте нуклеотидных замен. Среди SNP, найденных в наборе чтений, максимальная чатота — 98%, минимальная — 0.4%. Диаграмма 4 описывает распределение частот SNP, найденных в наборе.
Аннотация SNP по базам данных GWAS и ClinVar описывает предполагаемую взаимосвязь между SNP и фенотипом. Такая связь была найдена с помощью базы данных GWAS для трех SNP: замены в позициях 11374866, 31095171 и 56969148 ассоциированы с чертами, связанными с ожирением, болезнью Паркинсона и метаболическим синдромом соответственно; в базе данных ClinVar найденные SNP не аннотированы.
Файл pivot.tsv содержит информацию, полученную из аннотации по всем БД, для каждого SNP.