Учебный сайт Сергея Маргасюка
Применение EMBOSS и сравнение геномов
Работа с нуклеотидными последовательностями средствами пакета EMBOSS
Опишем выполнение нескольких заданий с помощью программ пакета EMBOSS:
-
Задача: объединить несколько файлов в формате fasta в один;
Входные данные: file1.fasta, file2.fasta, listfile1.txt;
Команда: seqret @listfile1.txt task1.fasta -auto;
Выходные данные: task1.fasta;
-
Задача: файл в формате fasta с несколькими последовательностями разделить на отдельные fasta файлы;
Входные данные: file3.fasta;
Команда: seqretsplit file3.fasta -auto;
Выходные данные: ab10a_arath.fasta, ab10b_arath.fasta, ab10c_arath.fasta;
-
Задача: из файла с хромосомой в формате .gb вырезать три кодирующих последовательности по указанным координатам "от", "до", "ориентация" и сохранить в одном fasta файле;
Входные данные: file4.gb, listfile2.txt;
Команда:seqret @listfile2.txt task3.fasta -auto;
Выходные данные: task3.fasta;
-
Задача: транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода, результат - в одном fasta файле;
Входные данные: task3.fasta;
Команда:transeq task3.fasta task4.fasta -table 0 -auto;
Выходные данные: task4.fasta;
-
Задача: транслировать данную нуклеотидную последовательность в шести рамках;
Входные данные: file4.gb;
Команда:transeq file4.gb:[214567:217231] task5.fasta -table 0 -frame 6 -auto;
Выходные данные: task5.fasta;
-
Задача: перевести выравнивание из формата fasta в формат msf;
Входные данные: file5.fasta;
Команда:seqret fasta::file5.fasta msf::task6.msf -auto;
Выходные данные: task6.msf;
-
Задача:выдать в выходной поток число совпадающих букв между второй последовательностью выравнивания и всеми остальными (на выходе только имя последовательности и число);
Входные данные: task6.msf;
Команда:infoalign msf::task6.msf stdout -refseq 2 -only -name -idcount -auto;
Выходные данные:см. вставку 1;
Вставка 1: выходные данные задачи 7
FER_CAPAA_1-157 96
FER_CAPAN_1-145 144
FER1_SOLLC_1-145 115
Q93XJ9_SOLTU_1-145 115
FER1_PEA_1-150 87
Q7XA98_TRIPR_1-153 92
FER1_MESCR_1-149 93
FER1_SPIOL_1-148 85
FER3_RAPSA_1-157 68
FER1_ARATH_1-150 90
FER_BRANA_1-157 71
FER2_ARATH_1-150 86
Q93Z60_ARATH_1-150 66
FER1_MAIZE_1-150 79
O80429_MAIZE_1-142 72
-
Задача: перевести аннотации особенностей в записи формата .gb в табличный формат .gff;
Входные данные: task4.gb;
Команда: featcopy gb::file4.gb gff::task8.gff -auto ;
Выходные данные: task8.gff;
-
Задача: из данного файла с хромосомой в формате .gb получить fasta файл с кодирующими последовательностями и добавить в описание каждой последовательности функцию белка (из поля product);
Входные данные: task4.gb;
Команда: extractfeat gb::file4.gb fasta::task9.fasta -auto -type cds -describe product;
Выходные данные: task9.fasta;
-
Задача: перемешать буквы в данной нуклеотидной последовательности и проверить с помощью blastn сколько "достоверных" находок (с E-value < 0.1) найдется в нуклеотидном банке данных;
Входные данные: file6.fasta;
Команда: shuffle -o task10.fasta file6.fasta;
Выходные данные: task10.fasta, HitTable.txt; минимальный E-value найденной последовательности — 1.6;
-
Задача: найти частоты кодонов в данных кодирующих последовательностях;
Входные данные: task9.fasta;
Команда: cusp task9.fasta:BK006939_264_4097 task11.txt -auto;
Выходные данные: task11.txt;
-
Задача: найти частоты кодонов в данных кодирующих последовательностях частоты динуклеотидов в данной нуклеотидной последовательности и сравнить их с ожидаемыми;
Входные данные: task9.fasta;
Команда: compseq task9.fasta:BK006939_264_4097 task12.txt -word 2 -calcfreq -auto;
Выходные данные: task12.txt; для динуклеотидов AC, AG, CA, CT, GA, GC, TG, TT частота выше ожидаемой; отношение наблюдаемой и ожидаемой частот не превосходит 1.3;
Построение карты локального сходства геномов
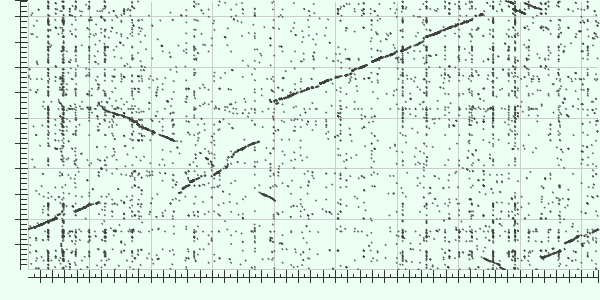
При выполнении данного задания с помощью blastn suite-2sequences был построен набор локальных выравниваний хромосом геномов Escherichia coli, штамм K-12 (ID NC_000913.3) и Enterobacter cloacae (ID NC_014121.1). О значительном сходстве геномов свидетельствует 69% и 61% покрытие геномов качественными выравниваниями соответственно. Внутри этих выравниваний 90% нуклеотидов идентичны. Соответствующая карта локального сходства приведена на рисунке 1.
Рисунок 1: карта локального сходства геномов
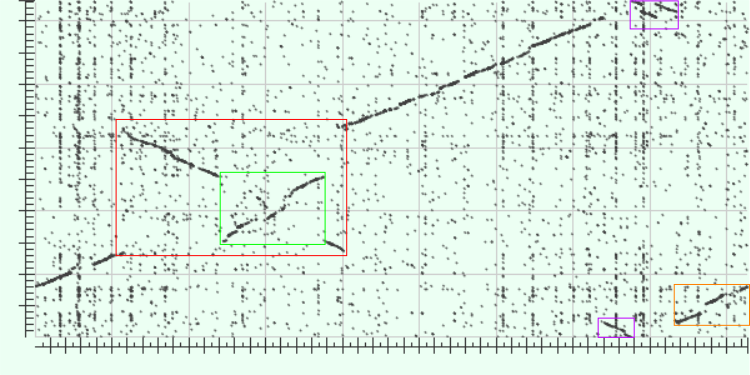
Отметим на данной карте некоторые участки, отклоняющиеся от наиболее крупной диагонали:
Рисунок 2: размеченная карта локального сходства геномов
Объясним отмеченные отклонения. Заметим, что участок, выделенный оранжевым, отклоняется от диагонали из-за неверного выбора начала отсчета на кольцевой хромосоме по одной из осей: например, если сдвинуть график вниз, помещая нижние части, выходящие за пределы графика, наверх, то этот участок окажется на диагонали. Аналогично, два фиолетовых участка должны быть расположены на графике рядом. Для отражения этих предположений о строении геномов была с помощью переноса участка снизу наверх построена новая карта (место соединения отмечено дополнительной линией сетки).
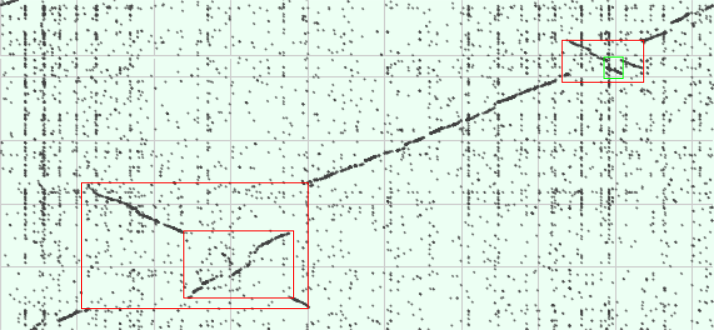
Рисунок 3: размеченная измененная карта локального сходства геномов
На новой карте отмечены красным случаи инверсии (переноса участка последовательности на комплементарную цепь), зеленым — случай транслокации.
© Сергей Маргасюк, 2015-2016