В практикуме 11 было показано, что на выравнивании Pfam значительно различаются две группы белков, соответствующих доменным архитектурам 1 и 2 соответственно: дерево делится на 2 клады, соответствующие почти точно архитектурам. Выделим одну из этих клад (соответствующую архитектуре 1) в гипотетическое подсемейство и построим соответствующий профиль. В Jalview получено (на основе выравнивания из Pfam) выравнивание этих генов; из него были удалены вручную участки с большим количеством гэпов (ссылка).
По этому выравниванию был построен профиль HMM.
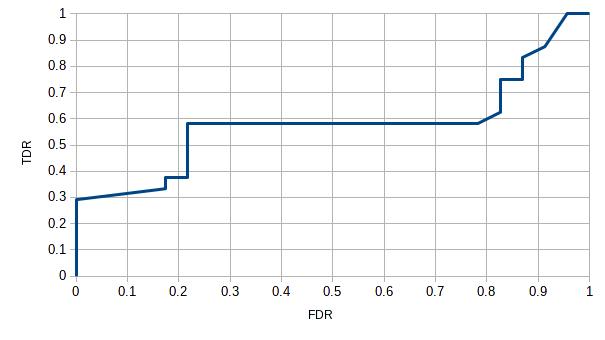
Выберем пороговое значение T. Для этого найдем T с помощью нашей HMM на файле subfamily1.fasta (последовательности принадлежат к подсемейству) и файле subfamily2.fasta (последовательности принадлежат к подсемейству). С помощью Excel (roc.ods)была получена кривая ROC для этой HMM.
График показывает, что (при отсутствии причин для предпочтения повышения TDR или понижения FDR) оптимальный выбор порога — на уровне TDR около 0.6 (T = 198, минмальный E-value находки = 3.4e-58).
Применим HMM с найденным порогом ко всем генам из выравнивания Pfam, содержащим домен Maelstrom. Часть выдачи (список генов) была помещена в скрипт на R (1.R) и сравнена со списком всех генов с архитектурой 1 из практикума 11: из 68 найденных HMM генов 25 имеют архитектуру 1. Чувствительность HMM 21%, специфичность 58%
| в подсемействе | вне подсемейства | сумма | |
| выше порога | 25 | 43 | 68 |
| ниже порога | 81 | 61 | 152 |
| сумма | 116 | 104 | 220 |
У метода недостаточная чувствительность при достаточно высокой специфичности: многие гены из подсемейства не были найдены.