Знакомство с BLAST
Задание 1. Нахождение гомологов моего белка(Краситель-обесцвечивающей пероксидазы Tfu_3078) в БД Swissprot
Описание параметров, включая Algorithm parameters, которые я использовали при запуске сервиса BLAST.
- Database - выбор базы данных, по которой будет произведён поиск белков, предположительно гомологичных
исходному(Query Sequence). На выбор предложены такие базы данных, как, например, UniProtKB/Swiss-prot(была выбрана мной), PDB, PAT, RefSeq и другие.
- Organism - позволяет сузить поиск гомологичных белков до белков в протеоме ввёденного организма/организмов или целого таксона.
(я не использовала данный параметр, однако не будет лишним о нём упомянуть)
- Algorithm. Начнём из далека. BLAST делится на 5 основных групп (Нуклеотидная, белковая, транслирующая,геномная, специальная).
В каждой из этих групп существуют собственные алгоритмы поиска. Мы используем "белковый" BLAST, предлагающий такие алгоритмы,
как BLASTP(медленное сравнение с целью поиска всех сходных последовательностей), PSI-BLAST (ищет последовательности с незначительным сходством),
PHI-BLAST и DELTA-BLAST (поиск белков, содержащих определённый паттерн или домен). В моём исследовании был примёнен алгоритм BLASTP.
- Max target sequences - выбираем, какое количесво последовательностей, удовлетворяющих введённым параметрам, появится у нас на экране.
Таким образом мы можем получить меньше находок, чем есть на самом деле. Мною было выставлено ограничение в 20,000 находок.
- Expect threshold - устанавливает ограничение на значение E-value. Порог, равный 10, устанавливается по умолчанию.
Что же такое E-value? Строго говоря, это математическое ожидание
числа находок BLAST с таким же или большим весом. А если говорить более простым языком, то E-value это параметр,показывающий,
стоит ли ожидать выравнивания с тем же или большим весом среди случайных последовательностей гипотетической базы данных(только она должна иметь
тот же размер, что и база даных, по которой мы осуществляем поиск). Таким образом, логично, что чем больше значение этого параметра, тем более
случайной может оказаться находка. То есть высокие значения E-value ставят под сомнения гомологичность исходного и найденного белка.
- Word size - параметр, задающий длину кусочков исходной последовательности для которых алгоритмы BLAST будут искать гомологичные куски
из базы данных. То есть Blast разбивает исходную последовательность на фрагменты заданной длины (6 по умолчанию)
и сравнивает их с фрагментами той же длины последовательностей, взятых из указанной базы данных. Выставляется какой-то порог веса выравнивания
фрагментов, ниже которого слова не считаются похожими. Таким образом программа BLAST создаёт словарь, где записано
каждое "слово" введённой последовательности(из 3-ёх символов, например), а напротив него разные белки, у которых что-то с ним выровнялось
(при этом вес выравнивания выше порогового). Далее программа начинает выравнивать последовательность по обе стороны от каждого слова на расстоянии
ниже определённого значения(порога). Постепенно по обе стороны от "слова" начинают появляться как новые совпадения, повышающие вес выравнивания,
так и гепы, за которые полагаются штрафы. Механизм остановки вырвнивания, основанного на данном алгоритме, довольно сложный.
- Matrix - выбор матрицы сравнения аминокислотных замен, от которого будет зависеть вес выравнивания. По умолчанию задана матрица BLOSUM62.
Матрица весов замен BLOSUM62 имеет порог кластеризации 62% и она ориентирована на цитоплазматические белки.
- Gap Costs - задаёт штраф за открытие инделя(extension:1) и за каждый последующий символ гепа(existence:11) в локальном выравнивании.
Этот параметр также влияет на вес выравнивания.
-
Полная таблица находок по заданным параметрам в BLAST
Далее из этой таблице мною были выбраны 6 последовательностей: 4 предположительно гомологичные(с хорошим покрытием и низким E-value)
и 2 предположительно негомологичные( с E-value 0.001 и 4.1 ).Более того, я старалась выбирать разнообразные по названиям белки.
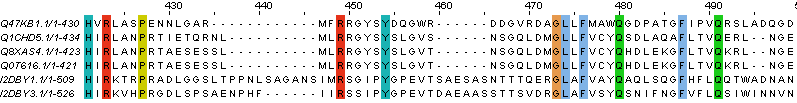 Рис.1 Множественное выравнивание 6-ти белков, построенное с помощью Jalview
Рис.1 Множественное выравнивание 6-ти белков, построенное с помощью Jalview
Из полученного множественного выравнивания нельзя сказать, что все белки в нём гомологичны друг другу.
Поэтому я решила убрать из него 2 последовательности, гомологичность которых изначально ставилась под сомнение.
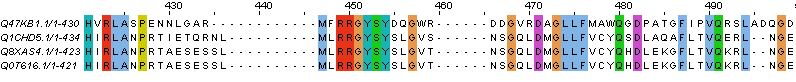 Рис.2 Множественное выравнивание гомологичных белков, построенное с помощью Jalview
Подтверждение гомологичности всех последовательностей:
Рис.2 Множественное выравнивание гомологичных белков, построенное с помощью Jalview
Подтверждение гомологичности всех последовательностей:
- На Рис.2 мы можем наблюдать участок выравнивания с 447 по 457 позиции без гепов, с высокой плотность консервативных позиций
по длине всего выравнивания. Более того он начинается и завершается абсолютно консервативной позицией.
Задание 2. Объяснение карт сходства двух белков
Для выполнения данного задания я выбрала два идентификатора записи Uniprot из разных групп:K1VG04_TRIAC и A0A163IMY9_DIDRA.
Далее я построила карту локального сходства, отметив опцию Align two or more sequences.
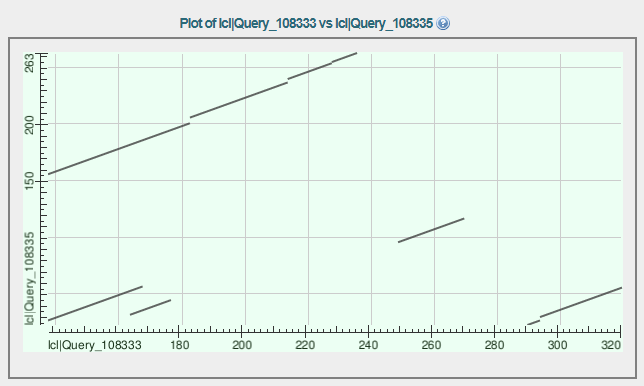 Рис.3 Карта локального сходства
Рис.3 Карта локального сходства
Между двумя белками программа BLAST построила 5 локальных выравниваний.
Четыре из которых можно считать случайными из-за высокого значения E-value. То есть далее я буду рассматривать только первое локальное выравнивание
(самая длинная полоса на Рис.3).
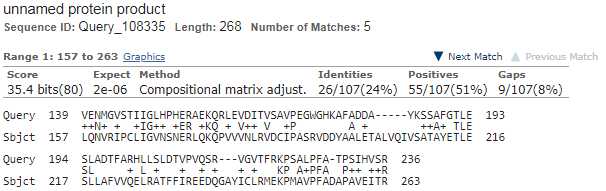 Рис.4 Достоверное локальное выравнивание (первое)
Выводы, сделанные из карты сходства(Рис.3) и локального выравнивания(Рис.4):
Рис.4 Достоверное локальное выравнивание (первое)
Выводы, сделанные из карты сходства(Рис.3) и локального выравнивания(Рис.4):
- Линия на карте сходства прерывается в трёх местах. Эти разрывы появляются в результате делеции аминокислотных остатков в
последовательности белка K1VG04_TRIAC.
- Мы видим, что последовательности выравниваются не целиком т.к. линия(диагональ) начинается не в нижнем левом углу,
а заканчивается не в верхнем правом. В локальном выравнивании присутствуют только части белковых последовательностей. На Рис.4 можно посмотреться,
какие а.о. вошли в выравнивание.
- Думаю, эти белки нельзя считать гомологичными, ведь процент иденичности в локальном выравнивании составляет всего 24%.
Задание 3. Игры с BLAST
Для игры с BLAST я взяла последовательность, которая точно не кодирует белок: "WE ARE PLAYING with BLAST". Выполняя поиск первый раз,
я оставила все параметры по умолчанию, кроме базы данных. Я искала гомологов для моей фразы среди последовательностей базы данных Swiss-Prot.
По моему запросу было найдено 7 последовательностей. Самый низкий E-value составил 0.041, что недостаточно для того, чтобы говорить о гомологичности.
Получается, BLAST не проведёшь, E-value говорит нам о том, что все произведённые выравнивания с высокой вероятностью являются случайными.
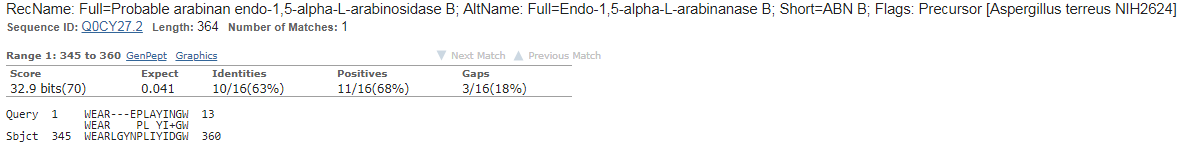 Наиболее удачное выравнивание с E-value 0.041 и высоким процентом идентичности/схожести, разве что короткое
Наиболее удачное выравнивание с E-value 0.041 и высоким процентом идентичности/схожести, разве что короткое
К сожалению, изменение параметров BLAST для воображаемого белка не приводило ни к каим изменениям находок.
Это довольно-таки странно. Но как уж есть.
Для дальнейшей игры я использовала последовательности своего белка(DYP_THEFY) в Swiss-Prot.
При первом запуске я вновь пользовалась параметрами по умолчанию, изменив лишь базу данных. Этот поиск был произведен для того, чтобы в дальнейшем
поле изменений параметров полученные данные было с чем сравнивать. Итак, во время первого запуска мне посчастливилось найти 13 белков (12 из них -
предположительные гомологи)
Посмотрим, как изменится их количество, если выставить word size, равный 2-ум. Думаю, число находок должно уменьшиться,
ведь при уменьшении этого параметра алгоритм BLAST, если можно так сказать, становитяс строже. Однако ,к моему удивлению, напротив, программа
выдала большее число находок - 25 (тем не менее по прежнему только у 12-ти из них хорошие значения E-value)
Далее я попробовала изменить матрицу весов замен на BLOSUM90 с более высоким порогом кластеризации. В результате я получила примерно те же
выравнивания, но с чуть большим весом. Таким образм, изменение данного параметра не влечёт за собой появление новых выравниваний.