Анализ транскриптомов. Bedtools.
Для выполнения практикума мне была предложена референсная последовательность 22-ой хромосомы человека.
Также были выданы чтения транскриптома, картирующиеся на участок 22-ой хромосомы человека. Чтения в формате fastq лежат в файле.
В ходе выполнения работы я использовала несколько команд, подробнее об их синтаксисе и выдаче в таблице:
| Команда | Выдача |
| Анализ качества чтений: | |
| fastqc infile.fastq | Команда производит контроль качества чтений, выдаёт отчёт в виде файла, формата .html |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 infile.fastq outfile.fastq TRAILING:20 MINLEN:50 | Таким образом я произвожу очистку чтений,
отрезая с конца каждого чтения нуклеотиды с качеством ниже 20, оставляя только чтения длиной не меньше 50 нуклеотидов.
Выдача команды - файл trimmed_chr22.fastq В дальнейшем к триммированным чтениям я применяю команду fastqc для проверки их качества. |
| Картирование чтений: | |
| hisat2-build chrX.fasta chrX | Выдача состоит из ряда файлов chrX.n.ht2 с проиндексированной референсной последовательностью |
| hisat2 -x chr22 -U infile.fastq -S outfile.sam --no-softclip | Производится картирование триммированных
чтений на проиндексированную последовательность.
Параметр --no-spliced-alignment был убран, дело в том, что мы работаем с транкриптомом, получается, из предоставленных чтений были вырезаны интроны в процессе сплайсинга, значит нужно разрешить картировать их на геном с разрывами. -x: начало названий нескольких проиндексированных файлов (их около восьми) -U: файл с чтениями -S: файл выдачи Выдача команды - файл mapped.sam |
| samtools view -b infile.sam -o outfile.bam | Перевод выравнивания
чтений с референсом в бинарный формат .bam.
-b переводит формат .sam в .bam -o позволяет ввести название файла для выдачи Выдача команды - файл mapped.bam |
| samtools sort infile.bam outfile_sorted | Сортировка выравнивания по
координате в референсе
Опции отсутствуют Формат аутфайла дописывать не надо, команда автоматически припишет .bam Выдача команды - файл mapped_sorted.bam |
| samtools index sorted_infile.bam | Индексирование отсортированного файла
Опции отсутствуют Название выводного файла вводить не следует, тк команда сама приписывает формат .bai к входному файлу Выдача команды - файл mapped_sorted.bam.bai |
| Подсчёт чтений: | |
| htseq-count -f bam -s no -i gene_id -m union mapped_sorted.bam /nfs/srv/databases/ngs/Human/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf | grep -wv 0 >> annotation.txt |
Команда htseq-count занимается подсчётом чтений, попавших на разные участки хромосомы. Команда grep с параметрами -w -v выбирает строки без нулей из выдачи
htseq-count, ведь нас не интересуют гены, на котрые не картировались чтения
Общий вид команды htseq-count [options] alignment_file gff_file -f принимает формат .bam или .sam -s описывает направление цепи, по которой были выровняны риды (принимает варианты: no, yes, reverse - нет направления, прямое, обратное) -i GFF-атрибут, используемый в качестве feature ID -m Определяет, как программа будет интерпретировать положение прочтения относительно референсных генов - какое положение считать перекрыванием, а какое - нет. Параметр можно оставить по умолчанию Выдача команды - файл annotation.txt |
Оправданность триммирования:
В первичном файле находятся 24,294 чтения с длиной 25-51 пар нуклеотид. После триммирования остаётся 23,459 чтений с длиной 50-51. При этом, если обратиться к рисункам 1,2, станет понятно, что качество чтений еще до очистки было приемлемым. Получается, процедура триммирования не совсем оправдана, мы могли бы работать с исходным файлом.
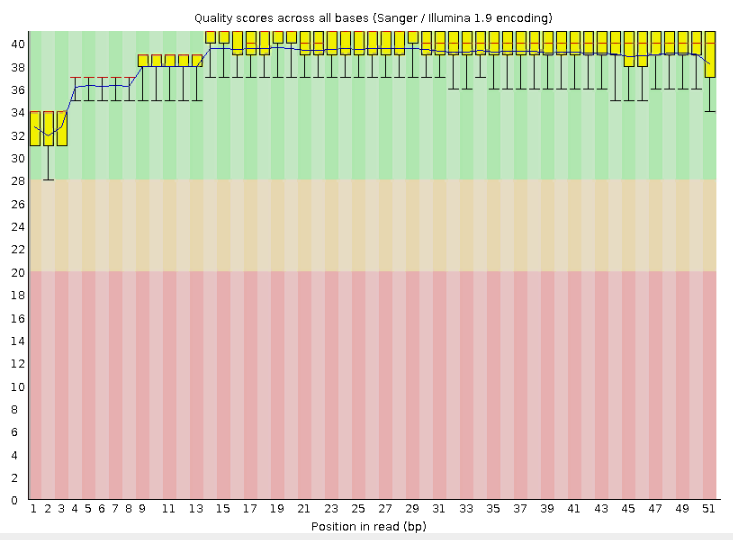
Рис.1 Чтения транскриптома до очистки
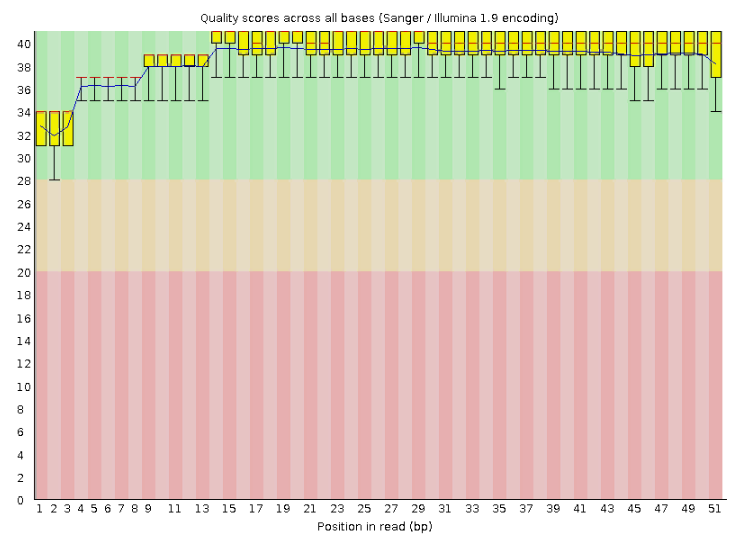
Рис.2 Чтения транскриптома после очистки
Анализ картирования чтений(выравнивания) на геном:
23459 reads; of these:
23459 (100.00%) were unpaired; of these:
366 (1.56%) aligned 0 times
23093 (98.44%) aligned exactly 1 time
0 (0.00%) aligned >1 times
98.44% overall alignment rate
Итого: всего 1,56% чтений не были картированы, в то время как 98,44% были сопоставлены с участком 22-ой хромосомы 1-2 раза. Можно с уверенностью сказать, что картирование прошло успешно.
Анализ результатов подсчёта чтений:
| Количество чтений | Какому участку хромосомы они принадлежат |
| 21,738 | ген ENSG00000185686.13 |
| 1,355 | Принадлежат учаткам хромосомы на стыке двух генов, соответственно их трудно отнести к какому-то конкретному гену(__no_feature) |
| 366 | Не принадлежат никакому участку хромосомы, тк это те чтения, которые по какой-то причине изначально не были картированы не 22-ую хромосому (__not_aligned) |
Большая часть чтений легла на ген ENSG00000185686.13. Даннй ген кодирует белки, находится на обратной цепи, чаще экспрессируется в клетках злокачественной опухоли - меланомы. Сложно судить о конкретных функциях гена, ведь синтезируемая с него мРНК, имеет около 16-ти вариантов альтернативного сплайсинга.