Сборка de novo
Передо мной стояла цель собрать часть генома бактерии Buchnera aphidicola без опоры на референсную последовательность. В качестве материала для работы мне предоставили код доступа проекта по секвенированию - SRR4240378. На странице проекта - http://www.ebi.ac.uk/ena/data/view/SRR4240378 я нашла ссылку для скачивания fastq-файла по протоколу FTP. Скачанный файл содержит короткие (длины 39) чтения, полученные по технологии Illumina
Команды, использованные в ходе выполнения работы:
| Команда | Выдача |
| Анализ качества чтений: | |
| fastqc infile.fastq | Команда производит контроль качества чтений, выдаёт отчёт в виде файла, формата .html |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 infile.fastq outfile.fastq ILLUMINACLIP:adapters.fasta:2:7:7 | Таким образом я произвожу очистку чтений, удаляя риды, которые являются остатками адаптеров. | java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 infile.fastq outfile.fastq TRAILING:20 MINLEN:32 | Вторая стадия очистки чтений включает в себя отрезание с конца каждого чтения нуклеотидов с качеством ниже 20. Более того, оставляем только чтения длиной не меньше 32 нуклеотидов. |
| du -sh infile.fastq | Команда выдаёт размер файлов |
| Сборка: | |
| velveth velveth31 31 -fastq -short infile.fastq | Команда подготавливает k-меры длины k=31
-short т.к. чтения короткие и непарные velveth31 - директрия с файлами, получившимися на выходе |
| velvetg velveth31 | Производит сборку на основе k-меров |
Подготовка чтений программой trimmomatic:
Чтения, которые были получены на секвенаторе Illumina, прежде всего необходимо очистить от адаптеров, коротких(то есть ридов, длиной менее 32ух нуклеотидов) и некачественных последовательностей. Для этого воспользуемся программой trimmomatic. До и после очистки я исследоваал риды на качетсво программой fastqc. Результаты её запуска приведены ниже в таблице и на рисунках 1-2.
| Этап работы | Количество чтений | Длина чтений | Размер файла |
| Исходный файл | 4.420.587 | 36 | 446M |
| После удаления адаптеров | 4.338.743 | 1-36 | 437M |
| После удаления адаперов и очистки по качеству, длине | 4.154.736 | 32-36 | 418M |
Оказалось, что 1.85 процентов последовательностей ридов были адаптерами.
После очистки по качетсву и длине было удалено 184.007 чтений. Судя по результатм, можно сказать, что триммирование чтений являлось необходимым этапом работы.
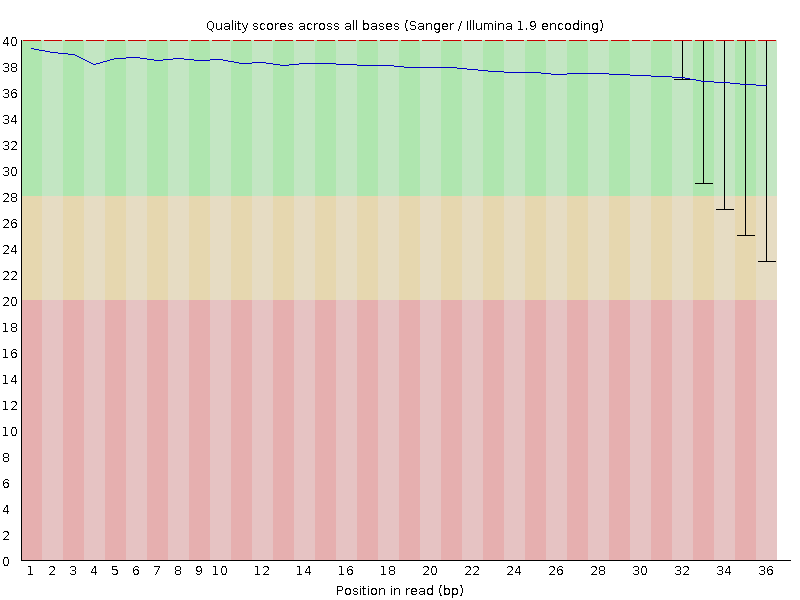
Рис.1 Качество чтений до и после удаления адаптеров (одинаковое)
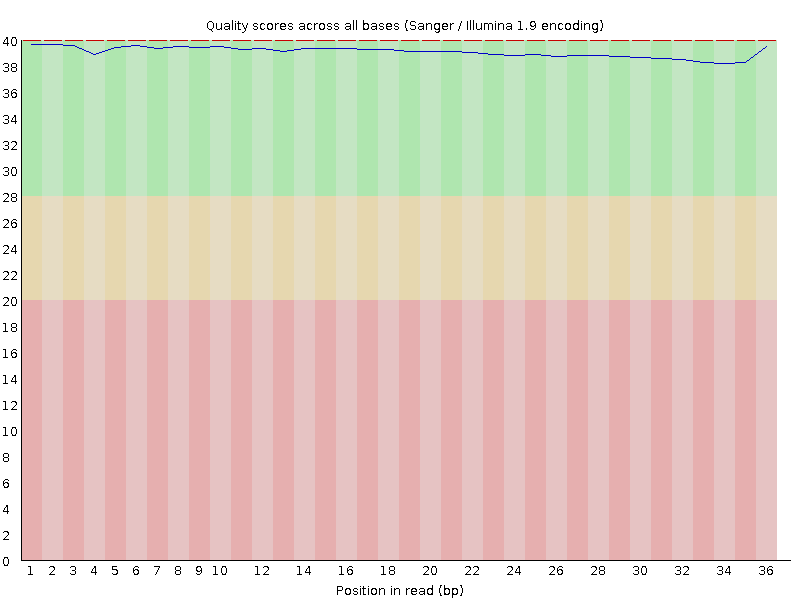
Рис.2 Качество чтений после очистки
Сборка:
Программа velveth подготавливает k-меры длины k=31. На их основе программа velvetg строит граф де Брёйна, собирая, таким образом, de novo. Сборка представляет из себя набор контигов, информацию о которых можно получить из файлов Log и stats.txt. Далее я приведу количественное описание сборки:
Получившийся граф содержит 351 узел.
N50 = 7.028
| Номера трёх самых длинных контигов | Длина | Покрытие |
| 8 | 36.746 | 20.017199 |
| 57 | 19.371 | 20.546642 |
| 15 | 16.745 | 20.901762 |
Вообше, среднее покрытие контигов(точнее, медиана) равно примерно 18ти. Значение хорошее, но, как говорится, могло бы быть и лучше. В сборке присутствуют и контиги с аномальными показателями покрытия. Их значения отличаются от медианы более, чем в пять раз. Они описаны в таблице:
| Номера контига | Длина | Покрытие |
| 293 (аномально большое покрытие) | 1 | 148.170 | 351 (аномально маленькое покрытие) | 1 | 1 |
Построение выравнивания трёх самых длинных контигов с хромосомой Buchnera aphidicola:
Программой megablast были выравняны каждый из трёх самых длинных контигов с хромосомой Buchnera aphidicola. Для этого на странице BLAST NCBI я нашла окошко "Align two or more sequences" и отметила его. Последовательности конигов были получены из файла contigs.fa, а хромосому я указала с помощью AC — CP009253. Характеристики находок приведены в таблице ниже:
Контиг 8
| Координаты участка хромосомы | Число гэпов | Identity |
| 500.370 to 508.806 | 351 (4%) | 76% |
| 510.438 to 516.539 | 187 (2%) | 79% |
| 481.997 to 488.106 | 308 (4%) | 74% |
| 496.111 to 500.325 | 154 (3%) | 75% |
| 493,487 to 494.864 | 13 (0%) | 80% |
| 480.874 to 481.545 | 20 (2%) | 82% |
| 495.033 to 495.148 | 5 (4%) | 90% |
Для того, чтобы понять как контиг лёг на хромосому, необходимо проанализировать карту локального сходства:
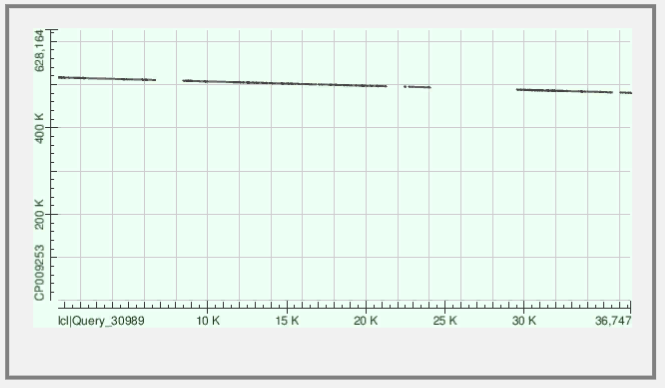
Рис.1 Контиг ложится на хромосому с разрывами - выравнились 7 частей контига и видно, что контиг лег на обратную цепь референса.
Контиг 15
| Координаты участка хромосомы | Число гэпов | Identity |
| 573.092 to 582.686 | 461 (4%) | 73% |
| 584.329 to 587.055 | 108 (3%) | 76% |
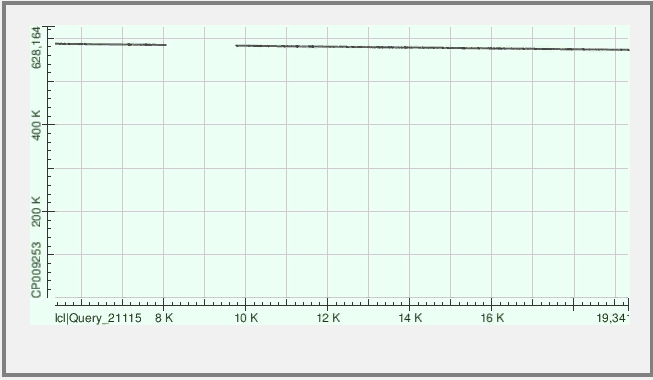
Рис.2 Контиг и хромосома имеют разное направление. Участков выравнивания - 2, то есть контиг ложится на хромосому практически непрерывно. Однако существенная часть контига не выравнивается вовсе.
Контиг 57
| Координаты участка хромосомы | Число гэпов | Identity |
| 144.368 to 151.796 | 243 (3%) | 78% |
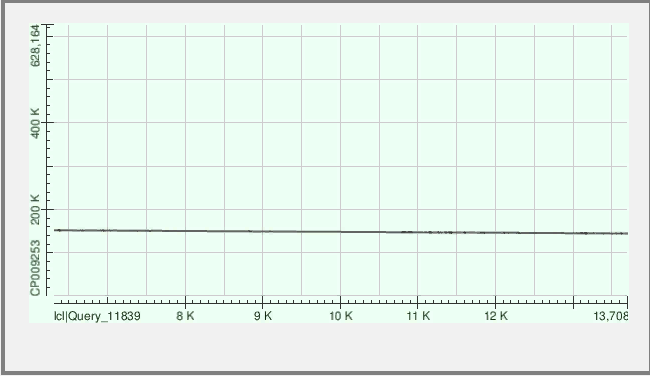
Рис.3 Контиг выравнивается с обраной цепью хромосомы без разрывов с хорошими показателями выравнивания. Но в выравнивании участвует не весь контиг, а только его часть.