Комплексы ДНК-белок
Задание 1: предсказание вторичной структуры заданной тРНК.
Упр.1. Предсказание вторичной структуры тРНК путем поиска инвертированных повторовЧтобы найти инвертированные участки в последовательности 1n77 тРНК я создала файл в формате fasta, содержащий ее нуклеотидную последовательность. Далее была использована команда einverted XXX.fasta с параметрами:
Gap penalty: 4
Minimum score threshold: 10/15/20
Match score: 3
Minimum score: -4
При этом, самый правдоподобный комплементарный участок был обнаружен при параметре Minimum score threshold = 15, он напоминает акцепторный стебель, найденный в предыдущем практикуме с помощью команды find_pair (pr2):
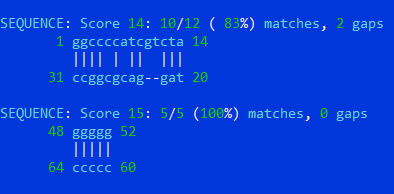
Когда был выставлен порог = 10, алгоритм попытался найти T-стебель и точнее предсказать акцепторный. По-моему, получилось некачественно.
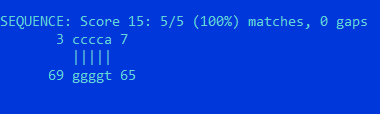
Если задать порог = 20, алгоритм результатов не выдает.
Упр.2. Предсказание вторичной структуры тРНК по алгоритму Зукера.С помощью сайта, на котором необходимо было всего лишь указать в окошке нуклеотидную последовательность тРНК, можно предсказать вторичную структуру 1n77. Алгоритм предложил две возможные структуры с отличиями в D-стебле, я выбрала ту из них, которая выглядит наиболее стабильной за счёт отсутствия основания без пары в D-стебле. К слову, другая сгенирированная модель имеет более низкую энергию, что могло бы говорить в её пользу, но эта разница незначительна(~1 kcal/mol).
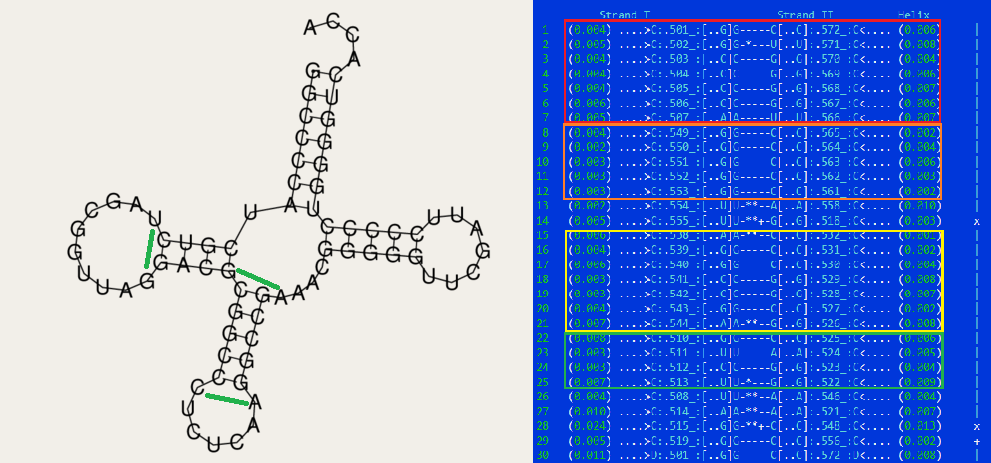
Как видно, акцепторный и Т-стебли полностью предсказаны алгоритмом Зукера. Однако в смоделиованном антикодоновом-стебле уже не хватает двух комплементарных связей, тех с которых начинается и заканчивается стебель реальной структуры. За счёт этого азотистые основания в D-стебле образуют водородную связь на одну пару раньше, что приводит к тому, что только 3 взаимодействия соответствуют результатам find_pair.
Чтобы подытожить результаты, полученные в Упр1 и 2, обратимся к таблице:| Участок структуры | Позиции в структуре(по результатам find_pair) | Результаты предсказания с помощью einverted (threshold 15) | Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | 5' 1 - 7 3'5' 66 - 72 3' | 5' 3 - 7 3' 5' 65 - 69 5'Буквенно предсказано 5 пар из 5 реальных, нумерация не совпадает почему-то | 7 пар оснований |
| D-стебель | 5' 10 - 13 3' 5' 22 - 25 3' | - | 5 пар оснований |
| T-стебель | 5' 49 - 53 3' 5' 61 - 65 3' | - | 5 пар оснований Исчезают 2 неканонические пары (отмеченные зелёными линиями: A-C, G-A) |
| Антикодоновый стебель | 5' 38 - 44 3' 5' 26 - 32 3' | - | 4 пары оснований Исчезает одна неканоническая(U-G), появляется одна новая (G-C) за счёт того что в предыдущем стебле гуанин не связывался с аргинином |
| Общее число канонических пар нуклеотидов | 23 | 5 | 21 |
Таким образои, можно уверенно сказать, что алгоритм Зукера моделирует вторичную структуру тРНК во много раз точнее, чем einverted.
Задание 2: Поиск ДНК-белковых контактов в заданной структуре
В данном задании я изучала ДНК-белковый комплекс с ID 1a02 (J и F цепи белка)
Упр.1. Определение групп атомов с помощью команды define в JMol.
Полярными считаются атомы кислорода и азота.
Неполярными - атомы углерода, фосфора и серы.
При этом назовем полярным контактом ситуацию, в которой расстояние между полярным атомом белка и полярным атомом ДНК меньше 3.5Å. Аналогично, неполярным контактом будем считать пару неполярных атомов на расстоянии меньше 4.5Å.
В таблице приведены результаты работы скрипта:| Контакты атомов белка с | Полярные | Неполярные | Всего |
| остатками 2'-дезоксирибозы | 3 | 7 | 10 |
| остатками фосфорной кислоты | 12 | 7 | 19 |
| остатками азотистых оснований со стороны большой бороздки | 3 | 15 | 18 |
| остатками азотистых оснований со стороны малой бороздки | 0 | 1 | 1 |
Изучив результаты, можно сделать вывод о том, что большиство связей ДНК формирует с остатками фосфорной кислоты, причём полярные среди них преобладают.
Упр.3. Схема ДНК-белковых контактов, полученная с помощью программы nucplot.Программа nucplot предназначена для визуализации контактов между ДНК и белком. Она работает только со старым форматом PDB (remediator 1a02.pdb > 1a02_old.pdb). Ввод (nucplot 1a02_old.pdb) позволяет получить на выходе файл nucplot.ps, который можно открыть с помощью программы gswin64.
Таким образом я получила следующее изображение:
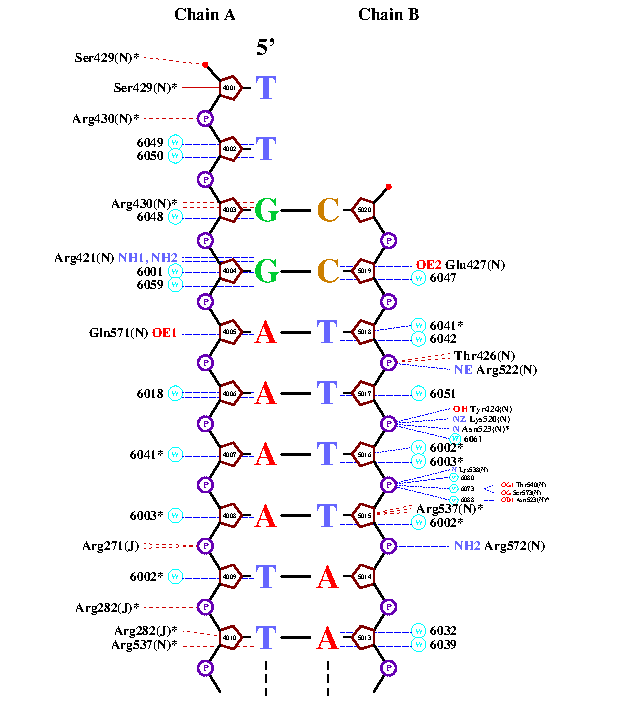
Упр.4.
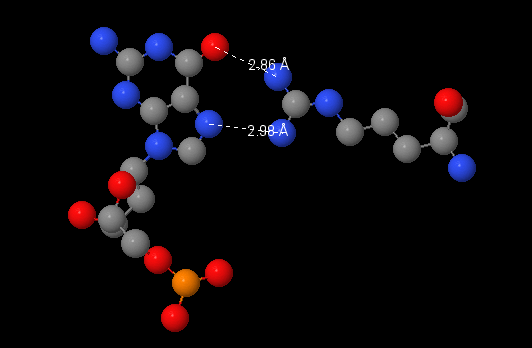
Здесь необходимо было выбрать аминокислотный остаток с наибольшим числом указанных на схеме контактов с ДНК. Я выбрала Arg537(N), который, судя по полученному с помощью nucplot изображению, формирует аж три связи с 2'дезоксирибозой ДНК. Эти связи отмечены коричневыми штришками, то есть они должны быть < 3.35 ангстрем. Однако в JMol команда (select within(3.35, [DT]5015) and [ARG]537) находит всего два атом, что противоречит полученной ранее информации. Интересно, где ошибка.
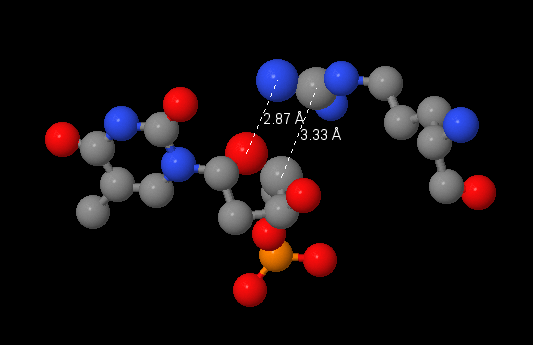
По-моему мнению наиболее важным для распознавания последовательности аминокислотным остатком является ARG421(N). Он образует две водородные связи с азотистым основанием, что является специфичным взаимодействием комплекса ДНК-белок.