Поиск по сходству (BLAST)
Задание 1: Определение таксономии и функции
нуклеотидной последовательности
Данная нуклеотидная последовательность была получена при анализе хроматограммы в пркатикуме 4.
Для выполнения задания использовался нуклеотидный бласт BLASTN, база данных Nucleotide collection. Алгоритм был запущен с параметрами по умолчанию. Выдача BLASTN оказалась следующей(приведены первые 10 результатов поиска):
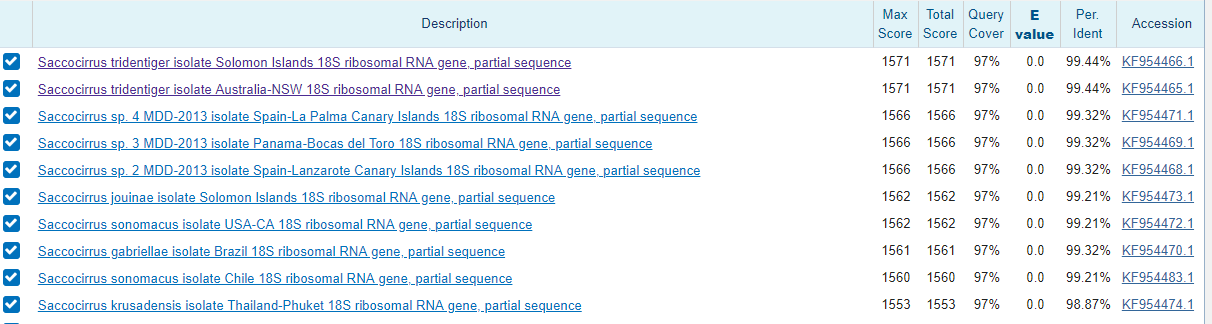
Предполагаемя функция - кодирование 18S рРНК тк находки с самыми высокими процентами идентичности (гены, одним из которых является исследуемая последовательность) отвечают именно за кодирование 18S рРНК.
Таксономию можно определить до вида - Pharyngocirrus tridentiger. Вывод сделан на основе того факта, что исследуемая последовательность лучше всего выравнивается с двумя органимами данного вида (первый был найден на Соломоновых островах, второй в Австралии). В связи с отсутствием затруднений в определении таксономии выравнвания я не привожу.
Задание 2: Сравнение списков находок нуклеотидных последовательностей тремя разными вариантами blast
Находки для последовательности из задания 1:
Чтобы нк ждать завершения алгоритма слишком долго, поиск был ограничен до рода Aricidea (taxid:273016).
| Алгоритм | Параметры | Число находок | Top score | Bottom score | Top Query Cover | Bottom Query Cover | Top Ident | Bottom Ident | Top E-Value | Bottom E-Value |
| megablast | Длина слова = 28 Match/Mismatch scores 1, -2 | 31 | 1408 | 1254 | 99% | 90% | 94.7% | 91.55% | 0.0 | 0.0 |
| blastn с параметрами по умолчанию | Длина слова = 11 Match/Mismatch scores 2, -3 | 32 | 1402 | 24.7 | 99% | 1% | 94.7% | 91.43% | 0.0 | 3.7 |
| blastn (с "чувствительными параметрами") | Устанавливаем чувствительные параметры:Длина слова = 7 Match/Mismatch scores 1, -4 Gapcosts 5,2 | 32 | 1308 | 26.4 | 97% | 1% | 94.93% | 91.62% | 0.0 | 1.2 |
На основании полученных результатов можно сделать вывод о том, что megablast выдаёт высоко сходные последовательности, те, которые ближе всего к исходной. Среди находок megablast отсутствуют недостоверные (e-valye для всех равен 0.0) Таким образом, он подходит для поиска близкородственных последовательностей. Его несомненным преимущетсвом является быстрый поиск. В результате работы blastn default и blastn sensitive поаявляется одна новая последовательность (DQ790052.1), она недостоверна, её можно заметить на Рис2 и Рис3. Это не должно удивлять, ведь blastn производит сравнение с целью поиска всех сходных последовательностей, а не только самых удачных. В связи с этим работает данный алгоритм немного медленнее предыдущего. К сожалению, у меня не получилось подобрать "чувствительные" параметры так, чтобы менялось кол-во находок, изменяются только их параметры, что абсолюно естественно, тк я, например, выставляю для данного алгоритма правила оценивания веса выравнивания 1,-4 .
 Рисунок 1 Выдача megablast
Рисунок 1 Выдача megablast
 Рис.2 Выдача blastn default .
Рис.2 Выдача blastn default .
 Рис.3 Выдача blastn sensitive
Рис.3 Выдача blastn sensitive
Находки для
CDS белка вируса Sulfolobales Beppu rod-shaped virus 1 clone D
| Алгоритм | Параметры | Число находок | Top score | Bottom score | Top Query Cover | Bottom Query Cover | Top Ident | Bottom Ident | Top E-Value | Bottom E-Value | Комментарии |
| megablast | Длина слова = 28 Match/Mismatch scores 1, -2 |
1 | 369 | 100% | 100% | 2e-98 | Находка единственная, она из вируса, из которого была взята последовательность CDS. | ||||
| blastn с параметрами по умолчанию | Длина слова = 11 Match/Mismatch scores 2, -3 | 99 | 347 | 48.2 | 100% | 15% | 100% | 73.15% | 1e-91 | 0.13 | Находок значительно больше, чем у megablast, однако около 10-ти из них недостоверны (e-value = 0.13) |
| blastn (с "чувствительными параметрами") | Устанавливаем чувствительные параметры:Длина слова = 7 Match/Mismatch scores 1, -4 Gapcosts 5,2 | 98 | 383 | 46.3 | 100% | 11% | 100% | 93.94% | 1e-102 | 0.49 | Находок примерно столько же, сколько и у обычного blastn, разница между этими двумя алгоритмами может быть прослежена при сравнении выравниваний Рис.4,5 |
 Рис.4 Выдача blastn default .
Рис.4 Выдача blastn default .
 Рис.5 Выдача blastn sensitive
Рис.5 Выдача blastn sensitive
Задание 3: Поиск гомологов трех белков в неаннотированном геноме
Необходимо было проверить наличие гомологов белков в геноме организма Amoboaphelidium protococarum (примитивный родственник грибов). Сборка генома лежит на kodomo в файле /P/y18/term3/block2/X5.fasta. Для выполнения задания мною были выбраны белки, которые наверняка должны присутствовать у всех эукариот:
Задание выполнялось с помощью локального BLAST+, установленного на kodomo. Для начала я создала локальную базу данных: makeblastdb -in X5.fasta -dbtype nucl. Параметр "-dbtype" определяет тип последовательности (у меня - нуклеотидная, следоватльно пишем nucl).
В ходе поиска гомологов был использвалалгоритм tblastn, который сравнивает изучаемую аминокислотную последовательность с транслированными последовательностями базы данных секвенированных нуклеиновых кислот. Команда для данного алгоритма выглядит так: tblastn -query xxx.fasta -db X5.fasta > xxx.out
Результаты:
CENPE_HUMAN

Score = 346 bits (887), Expect = 5e-95, Identities = 206/464 (44%), Positives = 276/464 (59%), Gaps = 56/464 (12%) Frame = +1
Query Cover = 15,6% (Данный параметр был посчитан вручную, я делила длину лучшего выравнивания на длину белка.)
Данные представлены для лучшей находки scaffold-100. Процент покрытия очень низкий, в выравнивании не видно гомологичных доменов. В связи с этим нельзя утверждать, что белок является гомологом. Интересно, с чем может быть связано его отсутствие в организме Amoboaphelidium protococarum.
TBB2_CAEEL
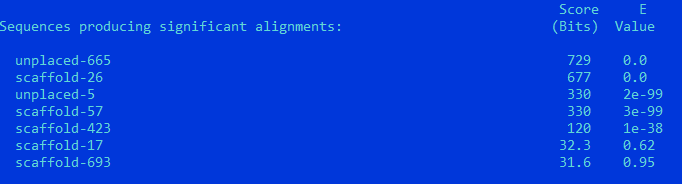
unplaced-665: Score = 729 bits (1882), Expect = 0.0, Identities = 371/455 (82%), Positives = 413/455 (91%), Gaps = 22/455 (5%) Frame = -2
Query Cover = 96%
Параметры сходства достаточно высокие, чтобы говорить о гомологии белков и сохранении функции.
RPB1_HUMAN
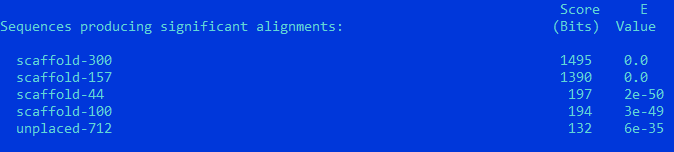
scaffold-300: Score = 1495 bits (3870), Expect = 0.0, Identities = 799/1547 (52%), Positives = 1079/1547 (70%), Gaps = 65/1547 (4%) Frame = +1
Query Cover = 76,6%
Предположительно, белки гомологичны, но параметры сходства не таковы, чтобы утверждать сохранение функции.
Задание 4: Поиск гена, кодирующего белок, в одном из контигов в геноме организма Amoboaphelidium protococarum.
Для начала я получила информацию о длинах контигов командой: infoseq X5.fasta -only -name -length
Затем мною был выбран scaffold-262 с длиной 48,647 пар нуклеотидов.
Последовательность данного скэффолда была получена так: seqret X5.fasta:scaffold-262 -out scaffold-262.fasta
Для поиска генов я использовала blastx, который переводит изучаемую нуклеотидную последовательность в кодируемые аминокислоты, а затем сравнивает её с имеющейся базой данных аминокислотных последовательностей белков. Поиск я ограничила царством Fungi, находки выбирала из RefSeq Protein.
Результаты поиска:

Вероятнее всего ген кодирует АТФ-зависимую РНК хеликазу.