Ресеквенирование. Поиск полиморфизмов у человека
Для выполнения практикума мне была предложена референсная последовательность 22-ой хромосомы человека.
Также были выданы чтения экзома, картирующиеся на участок 22-ой хромосомы человека. Одноконцевые чтения в формате fastq лежат в файле.
В ходе выполнения работы я использовала несколько команд, подробнее об их синтаксисе и выдаче в таблице:
| Команда | Выдача |
| hisat2-build chrX.fasta chrX | Выдача состоит из ряда файлов chrX.n.ht2 с проиндексированной референсной последовательностью |
| fastqc infile.fastq | Команда производит контроль качества чтений, выдаёт отчёт в виде файла, формата .html |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 infile.fastq outfile.fastq TRAILING:20 MINLEN:50 | Таким образом я произвожу очистку чтений,
отрезая с конца каждого чтения нуклеотиды с качеством ниже 20, оставляя только чтения длиной не меньше 50 нуклеотидов.
Выдача команды - файл cleared_chr22.fastq В дальнейшем к триммированным чтениям я применяю команду fastqc для проверки их качества. |
| hisat2 -x chrX -U cleared_chr22.fastq (файл с чтениями) -S outfile.sam --no-spliced-alignment --no-softclip | Производится картирование триммированных
чтений на проиндексированную последовательность.
Выдача команды - файл mapping.sam В связи с тем, что мы имеем дело с чтениями экзома, необходимо производить выравнивание по всему риду, более того в выравниваниях не должно быть инделей. |
| samtools view -b infile.sam -o outfile.bam | Перевод выравнивания
чтений с референсом в бинарный формат .bam.
Выдача команды - файл mapping.bam параметр -b переводит формат .sam в .bam; параметр -o позволяет ввести название файла для выдачи |
| samtools sort infile.bam soretd_outfile (формат аутфайла дописывать не надо, команда автоматически припишет .bam) | Сортировка выравнивания по
координате в референсе
Выдача команды - файл sorted_mapping.bam |
| samtools index sorted_infile.bam (Название выводного файла вводить не следует, тк команда сама приписывает формат .bai к входному файлу) | Индексирование отсортированного файла
Выдача команды - файл sorted_mapping.bam.bai |
| samtools mpileup -u -f chrX.fasta -g -o polymorph.bcf sorted_infile.bam | Создайте файл со
списком отличий(полиморфизмами) между референсом и чтениями в формате .bcf
Выдача команды - файл polymorph.bcf |
| bcftools call -cv polymorph.bcf -o polymorph.vcf | Создание файла со
списком отличий между референсом и чтениями в формате .vcf, с которым можно работать глазками, вотличие от бинарного .bcf файла
Выдача команды - файл polymorph.vcf Далее я буду использовать данный файл для описания трёх полиморфизмов |
| вручную удалила индели |
Файл - polymorph_without_indels.vcf |
| convert2annovar.pl -format vcf4 polymorph_without_indels.vcf > polymorph.avinput | Команда получает файл из .vcf файла,
с которым умеет работать программа annovar.
Выдача команды - файл polymorph.avinput |
| annotate_variation.pl -out refgene -build hg19 polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ | Aннотации файла с snp(polymorph.avinput) по базе данных refgene |
| annotate_variation.pl -filter -out dbsnp -build hg19 -dbtype snp138 polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ | Aннотации файла с snp(polymorph.avinput) по базе данных dbsnp |
| annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out 1000_genomes polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ | Aннотации файла с snp(polymorph.avinput) по базе данных 1000 genomes |
| annotate_variation.pl -regionanno -build hg19 -out gwas -dbtype gwasCatalog polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ | Aннотации файла с snp(polymorph.avinput) по базе данных GWAS |
| annotate_variation.pl polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ -filter -dbtype clinvar_20140211 -buildver hg19 -out clinvar | Aннотации файла с snp(polymorph.avinput) по базе данных Clinvar |
Задание1. Анализ качества чтений, прошедших процедуру триммирования:
Исходно было получено 11,427 чтений с длиной 30-100 пар оснований. После очистки чтений их осталось 11,091 с длиной от 50 до 100 нуклеотидов, как и требовалось. Сравнивая Рис.2 с Рис.1 можно чётко отследить улучшение качества чтений после триммирования. Эта процедура более чем оправдана, ведь в дальнейшем мы получим более точные данные.
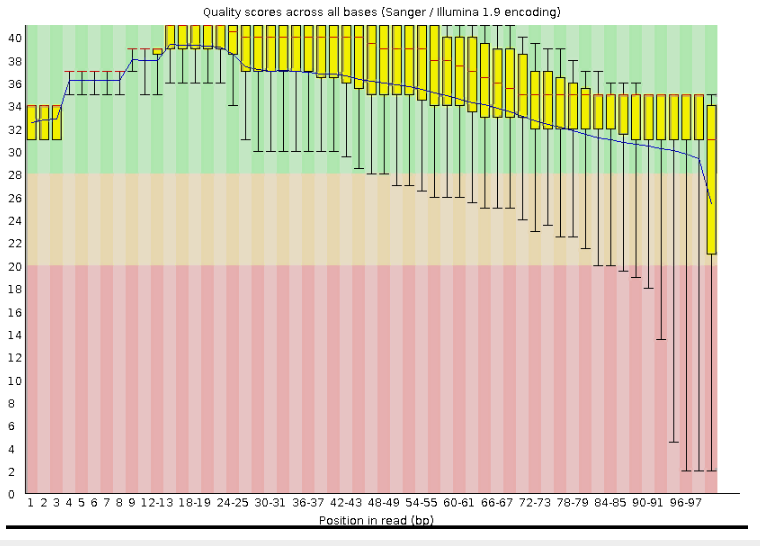
Рис.1 Оценка качества чтений до триммированиия (чистки)
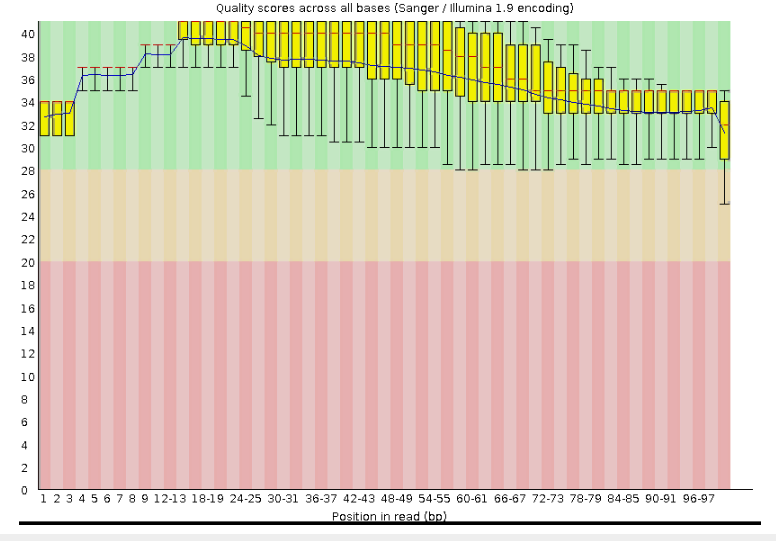
Рис.2 Оценка качества чтений после триммированиия
Задание2. Анализ картирования чтений на геном:
Информация, представленная ниже, выдаётся по результатам запуска программы картирования hisat2:11091 reads; of these:
11091 (100.00%) were unpaired; of these:
53 (0.48%) aligned 0 times
11026 (99.41%) aligned exactly 1 time
12 (0.11%) aligned >1 times
99.52% overall alignment rate
Получается, 53 чтения не удалось сопоставить с участком 22-ой хромосомы человека, 11,026 были картированы один раз, 12 дважды. Таким образом, на геном было картировано 99,52% чтений. Данные свидетельствуют о высоком качестве картирования.
Задание3. Поиск SNP и инделей (описание трёх полиморфизмов из файла) :
Всего было найдено 225 полиморфизмов, из которых 220 представляют из себя однонуклеотидную замену(snp), еще 5 - индели.
Среднее качетсво полиморфизмов: 67,26, медиана: 11,34. Я считаю, медиана даёт более точное представление о качестве полиморфизмов, среднее же "портится" немногочисленными высокими значениями.
Что касается покрытия, для него среднее значение: 25, медиана: 19. Вновь медиана лучше среднего характеризует покртыие. Покрытие, равное 19 можно считать хорошим.
| Координата | Тип полиморфизма | Референс | Чтение | Глубина покрытия данного места | Качество чтения в данном месте |
| 26159289 | Замена | G | A | 19 | 221.999 |
| 26219723 | Вставка | CTTTTTT | CTTTTTTT | 6 | 21.6211 |
| 26286738 | Замена | T | A | 62 | 225.009 |
Задание4. Аннотация SNP
Чтобы провести анализ полиморфизмов, необходимо было проаннотировать файл по различным базам данных. Было получено множество файлов с описанием однонуклеотидных замен.
В частноси, база данных refseq в annovar делит snp на несколько категорий в зависимости от того, в каком участке происходит замена. Данная информация взята из файла refgene.variant_function:
| Категория | Сколько snp попало в данную категорию |
| exonic | 26 |
| intronic | 186 |
| ncRNA_exonic | 1 |
| ncRNA_intronic | 6 |
Сложив все эти числа, получим общее количество snp - 219
Гены, в которые попали snp: TTC28, TTC28-AS1, APOL1, MYO18B
В файле с названием refgene.exonic_variant_function можно посмотреть, являются ли замены в экзонах (интроны не так интересны) значимыми. То есть там представлена информация о синонимичности и несинонимичности snp в экзонах. Итого: синонимичных замен 8, все остальные - несинонимичные.
В файле dbsnp.hg19_snp138_dropped лежат snp, у которых есть rs, всего их оказалось 177, оставшиеся 42 замены rs не имеют, следовательно в банке dbsnp они не описаны. Информацию о них можно найти в файле dbsnp.hg19_snp138_filtered
Можно сказать пару слов о частоте полученных замен, посмотрев в файл 1000_genomes.hg19_ALL.sites.2014_10_dropped. Cредняя частота состаляется 37%, но вновь среднее значение не так показательно, частота варьируется от 99% до 0,3%.
О клинической аннотации snp пишут в файле gwas.hg19_gwasCatalog, в базе данных clinvar ничего не получается найти.
Были найдены очень интерсные последствия однонуклеотидных замен:
gwasCatalog Name=Mathematical ability in children with dyslexia chr22 26159289 26159289 G A hom 221.999 19
gwasCatalog Name=Obesity chr22 28378472 28378472 A G hom 222.01 34
gwasCatalog Name=Glomerulosclerosis chr22 36661330 36661330 G A het 175.009 34
Я нашла эти замены по координатам в файле refgene.exonic_variant_function, чтобы посмотреть в каких генах происходит мутация. Попыталась понять, какая функция у этих генов, чтобы выстроить логическую цепочку между мутацией в конкретном гене и клиническими последствиями. Но, к сожалению, не смогла найти подробную информацию о функциях генов.