Секвенирование по Сэнгеру
1. Получить последовательность ДНК на основании данных, полученных из капиллярного секвенатора.
В этом задании глобально нужно было провести качественную расшифровку результатов секвенирования по Сэнгеру. Для этого необходимо было обработать две хроматограммы (с прямой и обратной цепочек), собрать из них контиг и разрешить все неоднозначности. Для этого были выданы файлы прямого и обратного прочтений. С обратного прочтения была получена перевернутая комплементарная цепь.
Все полученые файлы можно посмотреть здесь:
- 02_F.fasta
- 02_R.fasta
- 02_R_reverse_complement.fasta
- align.fasta
- align.needle
- Ссылка на полученное выравнивание
Ниже приведен результат выдачи программы Jalview. Он поможет определиться с консенсусной последовательностью.

Рис. 1. Полученное выравнивание в Jalview. Кликните на изображение, чтобы улучшить качество
Для анализа хроматограммы я использовала программу Unipro UGENE. По полученным данным можно сказать, что на прямом прочтении длины нечитаемых участков составляли 58 нуклеотидов в начале (c 5' конца) и 306 нуклеотидов в конце. На обратной цепи - 58 в начале и 252 в конце.
На глаз отношение сигнала к шуму составляет примерно 4:1. В целом хроматограмма хорошая, однако на концах имеются достаточно протяженные участки сильного шума. Неравномерность силы сигнала и шума представлена классическим образом - больше шума в начале и в конце прочтения.
Следующим шагом необходимо было установить неточности в прочтении, основываясь на данных хроматограммы и последовательности комплементарной цепи.
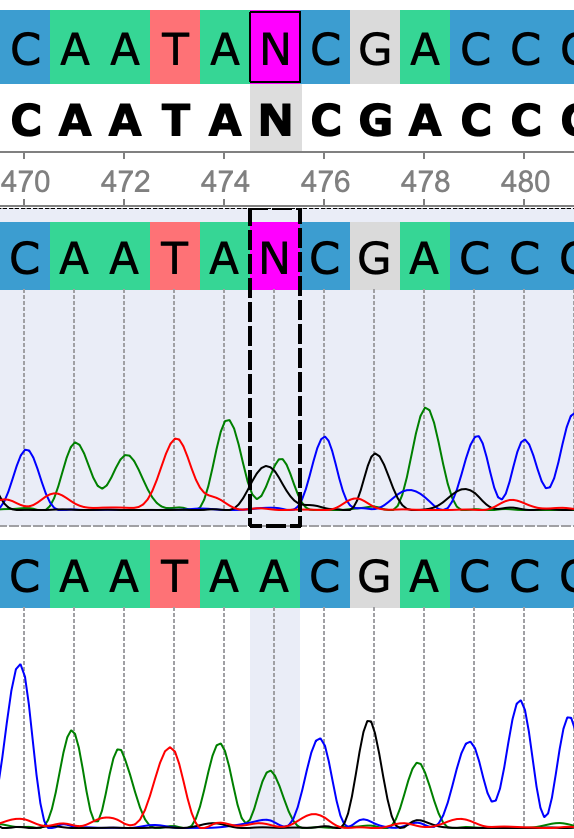

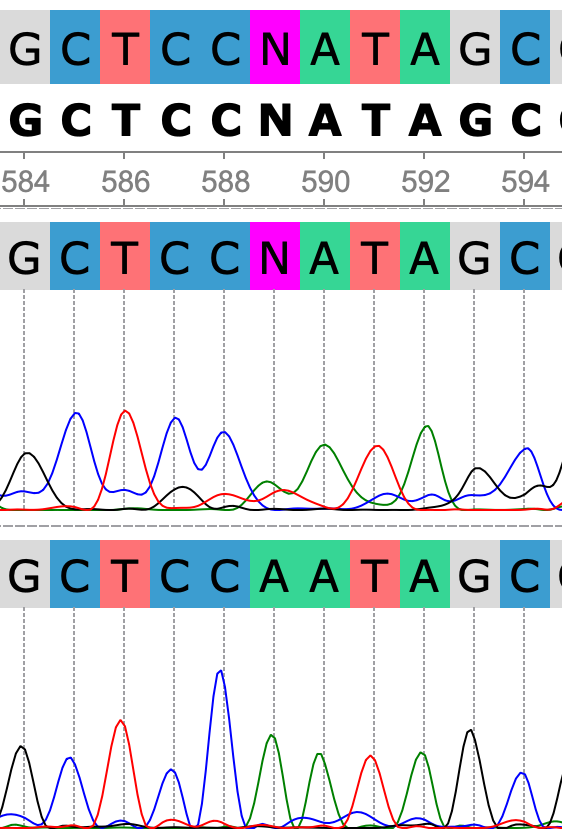
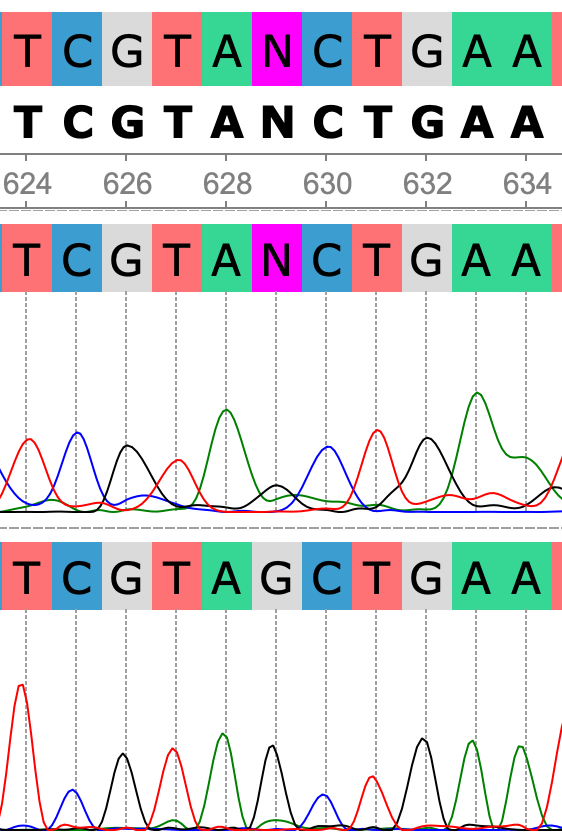
Далее из выравнивания скорректированных последовательностей был извлечен консенсус, который можно найти по ссылке.
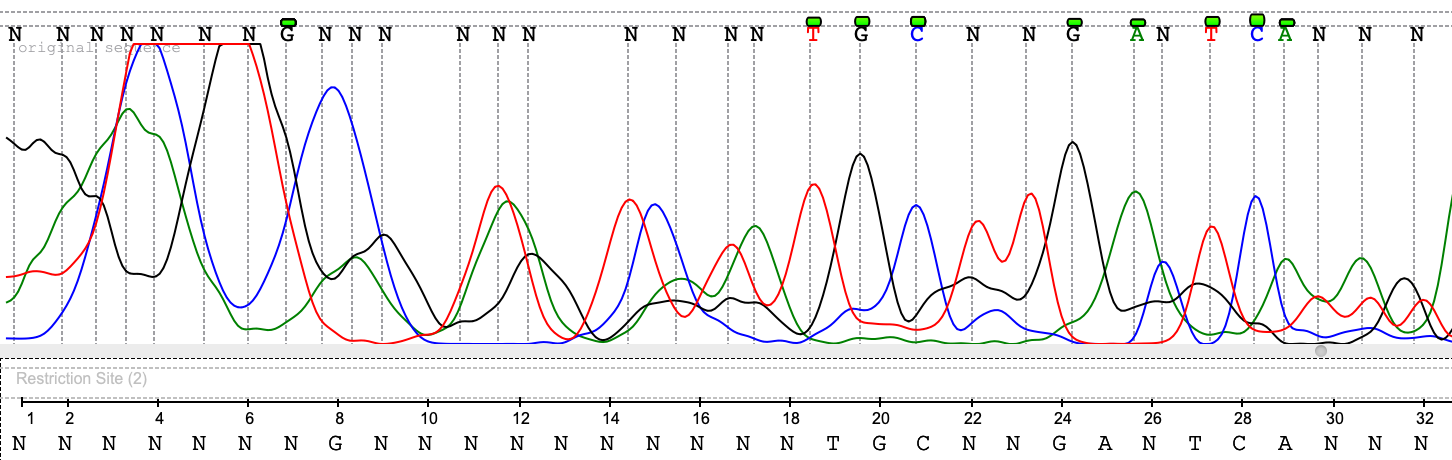
2. Нечитаемый фрагмент хроматограммы
Классический пример нечитаемых фрагментов - начало и конец хроматограммы. В качестве примера мною было выбрано начало хроматограммы прямого прочтения. На рисунке видны широкие, перекрывающиеся друг с другом пики, а также пятна краски. Это может быть связано с наличием коротких неспецифических участков, на которые отжигаются праймеры, а также с ошибками во время фореза.