
BLAST (Basic Local Alignment Search Tool) — это программа, использующая статистические методы для поиска нуклеотидных или аминокислотных последовательностей (subject sequences), похожих на искомую (query). Каждая найденная последовательность имеет набор параметров:
Description – описание последовательности
Max score – наибольший вес парного выравнивания
Total score – сумма весов всех возможных выравниваний
Query cover – процент покрытия исходной последовательности выравниванием
E value - математическое ожидание нахождения выравнивания с таким же или большим весом в случайном банке последовательностей. Для малых значений равно p value вероятности нахождения случайного выравнивания. Обычно находки с E < 0.05 считаются значимыми.
Ident - процент совпадающих аминокислот
Accession - идентификатор последовательности в GenBank
После запуска программы для белка с идентификатором NP_560184.1 Blast выдал следующие результаты:
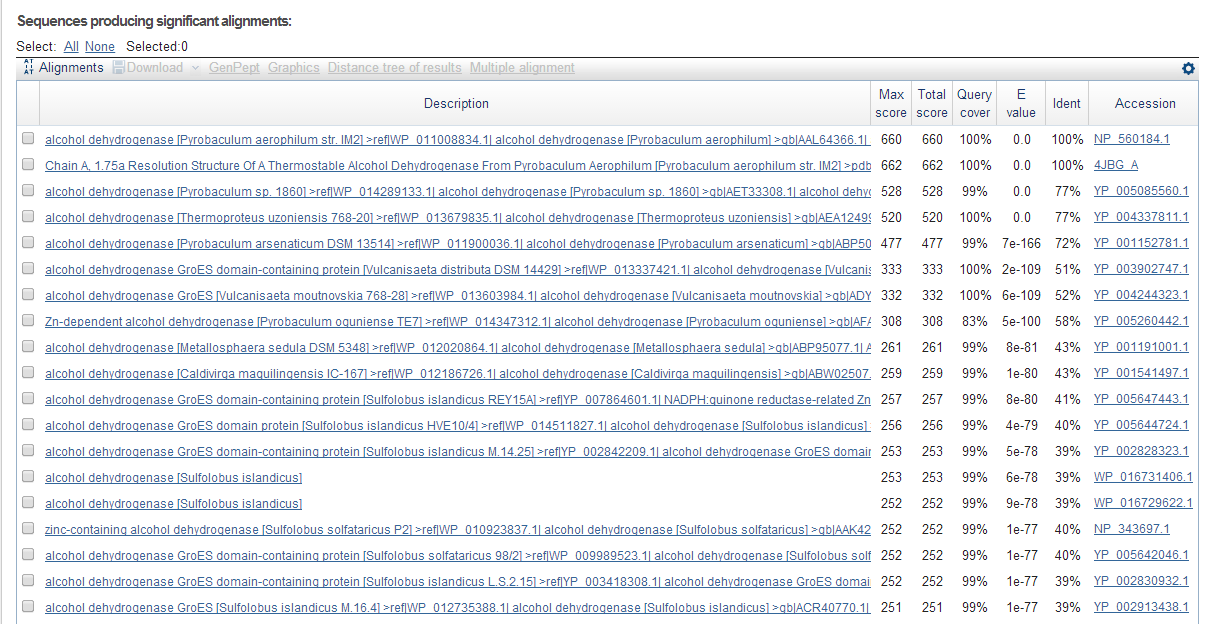
Рис.1 Первые 19 результатов работы программы
Все найденные последовательности имеют очень высокий процент покрытия и почти все являются алкоголь дегидрогеназами в различных прокариотических организмах. В качестве объекта для дальнейшего изучения я выбрала находку с идентификатором YP_005260442.1, Identity 58%, E value 5*e^(-100), Query coverage 83%, Identity 58%, Max score и Total score 308. Выравнивание этой и исходной последовательностей, выданное программой Blast вы можете увидеть на
В задании 2 было предложено построить парное выравнивание последовательности моего белка - NP_560184.1 и выбранного — YP_005260442.1 (см. рис 2) и построить карту локального сходства опять же с помощью программы Blast (см рис 3).
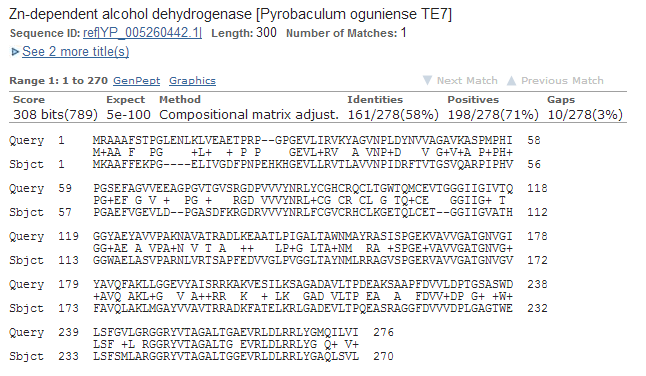
Рис. 2 Выравнивание исходной последовательности и найденной
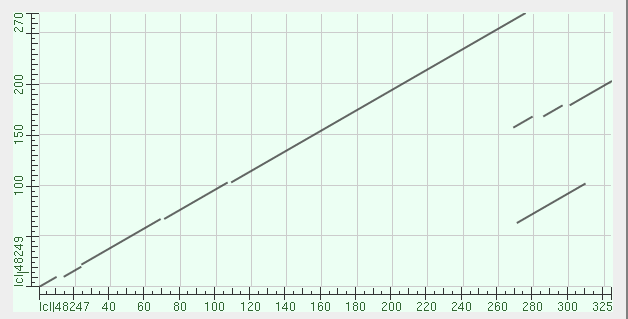
Рис.3 Карта локального сходства двух последовательностей. По горизонтали позиции в белке с идентификатором NP_560184.1, по вертикали YP_005260442.1
Из карты локального сходства видно, что у моего белка (query) есть участок в конце последовательности, выравнивающийся несколько раз на последовательность находки. Причем в двух других находках, которые я решила проверить, такого не наблюдается. (см. рис. 4 и рис.5). Логично было бы предположить, что это некие повторы в последовательности белка, но тогда похожая картина должна была наблюдаться и во всех остальных результатах, мы же такого не наблюдаем.
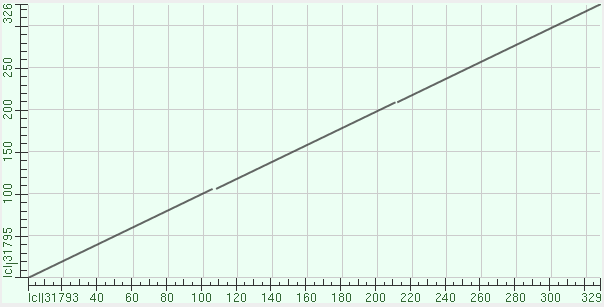
Рис. 4 Карта локального сходства двух последовательностей. По горизонтали позиции в белке с идентификатором NP_560184.1, по вертикали YP_001152781.1
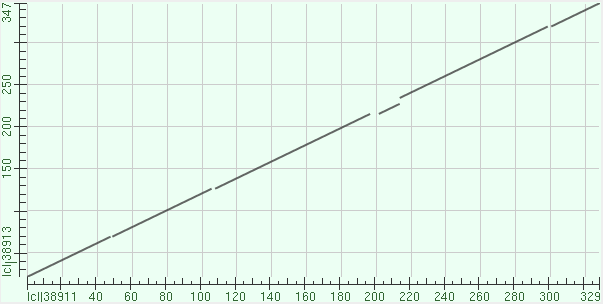
Рис. 5 Карта локального сходства двух последовательностей. По горизонтали позиции в белке с идентификатором NP_560184.1, по вертикали YP_001191001.1
Я затрудняюсь предложить хорошее и логичное объяснение данному явлению, но возможно это издержки алгоритма программы и эти участки схожи, но не гомологичны.
В задании 3 нужно было найти гомологов последовательности среди эукариотических организмов. Для выполнения задания, я изменила в параметрах программы базу данных для поиска на Uniprot/Swissprot и запуcтила Blast. Программа выдала 8 находок с E value менее e^(-17), что является очень хорошим показателем. Результат работы программы представлен на рис. 6
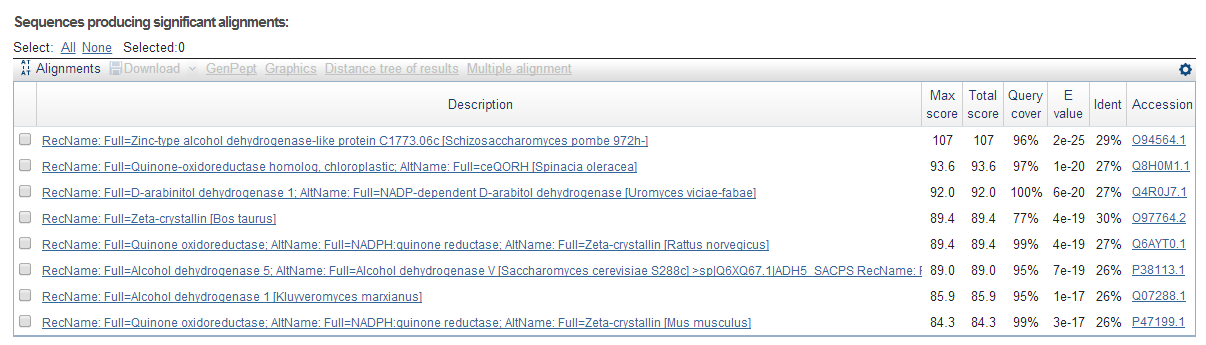
Рис. 6 Схожие с query последовательности из домена Eukaryota
Результат довольно логичен, т.к. мой белок — алкоголь дегидрогеназа, хотя и имеет значительные различия в механизме работы и строении активного центра у эукариот и прокариот, но имеют схожие функции и возможно общее происхождение с найденными белками (3 алкоголь дегидрогеназы, 4 хинон оксиредуктазы, 1 D-arabinitol дегидрогеназа).
Проект множественного выравнивания всех 8 найденных аминокислотных последовательностей можно скачать здесь, выравнивание -здесь.Абсолютно консервативных позиций 18 из 383 (4.7%), функционально консервативных 42 (10.9%). Это неплохой результат, т.к. вероятность нахождения одного абсолютно консервативного столбца, с учетом того, что в выравнивании 8 последовательностей крайне мала.