
Был выбран домен PF02574 -S-methyl_trans – домен, включающий в себя S-метилтрансферазы. Белки с данным доменом крайне важны для организма, у человека участвуют в развитии нервной и кровеносной системы, их недостаток вызывает тяжкие нарушения в развитии.
С помощью скрипта swisspfam-to-xls.py (python swisspfam-to-xls.py -z /srv/databases/pfam/swisspfam.gz -o DOMAINS_INFO.txt.xls -p PF02574), данного нам для выполнения данного практикума, были получена информация о всех доменах в последовательностях, имеющих наш домен. - DOMAINS_INFO.txt.xls
Затем с помощью сервиса Retrieve / Uniprot ID Mapping был получен файл в формате Uniprot, содержащий информацию о всех последователбностях - UNIPROT.txt
С помощью uniprot-to-taxonomy.py была получена таксономия для каждой последовательности ( к сожалению, некоторые последовательности были удалены/слиты/переименованы и Uniprot выдал информацию о них под новыми именами, поэтому для некоторых в дальнейшем таксономия оказалась неопределенной) - TAXONOMY.txt.xls
С помощью get_domain_arch.py были получены архитектуры всех последовательностей в удобном для восприятия формате и формате, удобном для работы последующих скриптов. - DOMAINS_INFO_PROCESSED.txt.xls
С помощью средств Excel в таблицу, представляющую собой результат работы предыдущего скрипта была добавлена таксономия (к какому домену принадлежит организм, у которой найден данный белок). Полученная модифицированная таблица - ARCH_TAX.txt.xls - была подана на вход скрипту get_arch_and_tax.py.
С помощью него была получена сводная информация о каждой архитектуре, для которой известно не менее 25 последовательностей-представителей (сколько в ней последовательностей, каким таксонам принадлежат) - ARCH_TAX.txt
В результате для дальнейшей работы были отобраны архитектура 2 , архитектура 3 и архитектура 6 из файла (скрипт был перезапущен – в файле теперь это 2, 5 и 3 архитектуры соотвественно)
Из них было выбрано по 10 представителей для доменов Eukariota и Bacteria (с помощью одного из результатов работы предыдущего скрипта)
С помощью choose.py были отобраны последовательности, сооотвествующие этим организмам из общего файла Pfam, содержащего последовательности выбранного домена, вырезанные из последовательностей белков - common.fasta
Далее они были выравнены с помощью сервиса Muscle - aln-fasta.fasta
Проект JalView - aln.jvp
Затем с помощью программы Mega методом Maximum Likelihood было построено дерево. Его изображение - res.png см. рис.1 Его файл – res.nwk
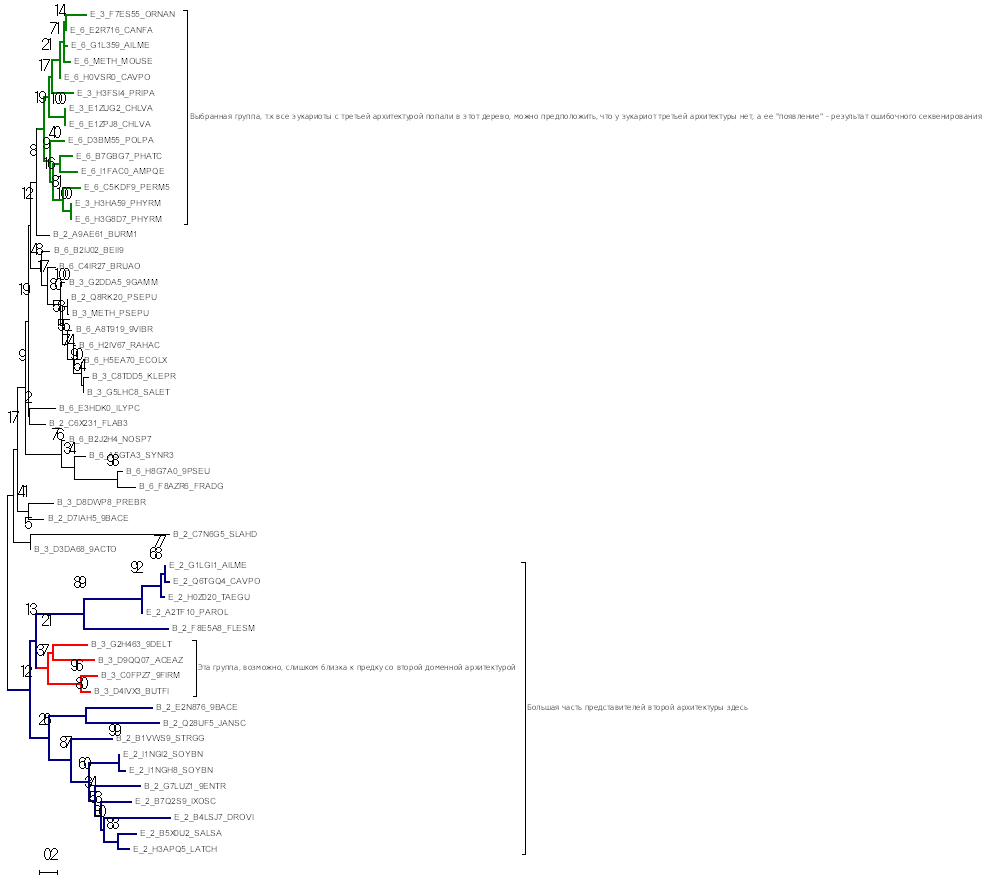
Рис.1. Дерево эволюции домена
Выделить какую-либо кладу, которая включала бы в себя все последовательности какой-то одной архитектуры и не включала последовательностей другой не получается.
Это можно объяснить либо низким уровнем мутаций в гене данного белка, либо (в случае к примеру E_3- ошибками секвенирования, т.к 3 и 6 архитектуры имеют два общих домена, 6 к тому же имеет еще 4 домена – возможно программа автоматическо разметки генома прочитала до стопкодона, полученного из-за ошибки секвенирования и потому не заметила следующих доменов). Особенно часты перестройки в невыделенной кладой, но тут опять же, это скорее всего объясняется низким уровнем мутаций
Далее для построения была выбрана группа, выделенная зеленом цветом – Эукариоты с 3 и 6 архитектурами.
С помощью hmm2build и hmm2calibrate была получена СММ для выбранных последовательностей - ch_model.hmm
C помощью hmm2search с использованием этого профиля был проведен поиск по всем последовательностям, содержащим выбранный домен - hmmsearch_out.txt
С помощью скрипта create_ROC_and_EP_v2.py была получена разметка правильных/неправильных предсказаний - res_predict_prove.txt.xls, посчитаны true positives, false positives, true negatives и false negatives для каждого порога e-value - res_roc.txt, построена roc-кривая см. рис. 2.
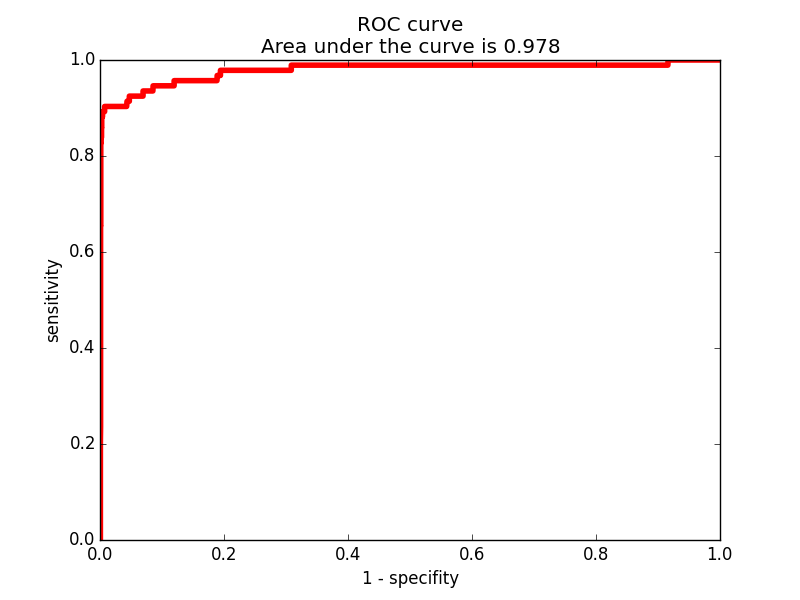
Рис.2. ROC кривая.
Из площади под кривой – 0.978 можно заключить, что данный профиль пригоден для использования.
Для выбранного порога e-value - Evalue = 4.2e-214
true_positives = 83
false_positives = 13
true_negatives = 3199
false_negatives = 10
Специфичность равна при выбранном пороге равна 0.996,
Чувствительность – 0.892
Обобщенные результыт приведены в таблице PFAM_RESULTS.txt.xls