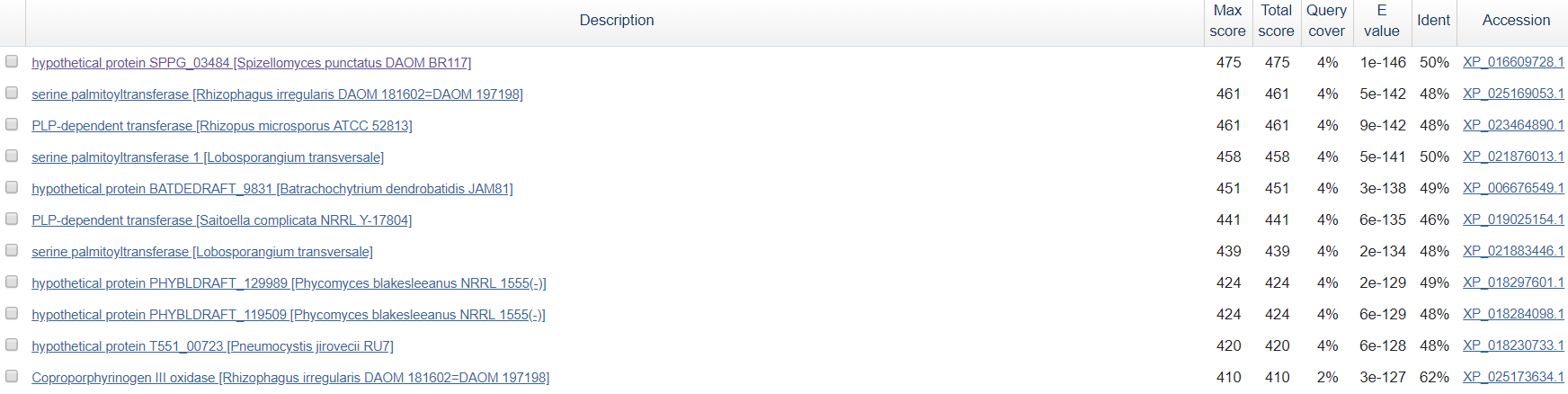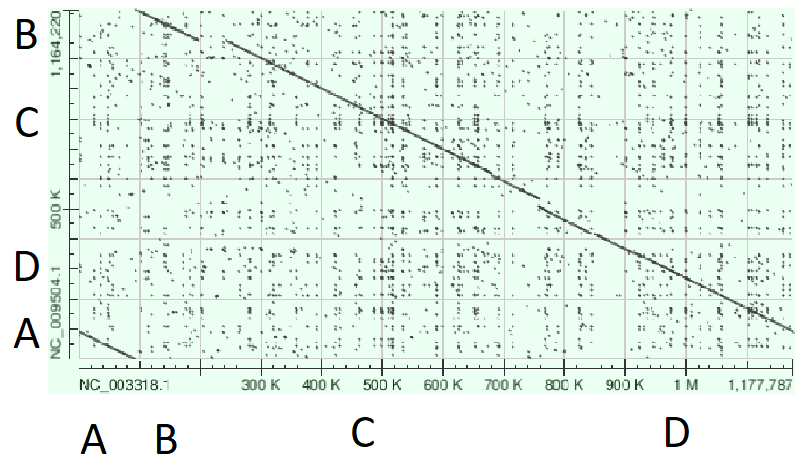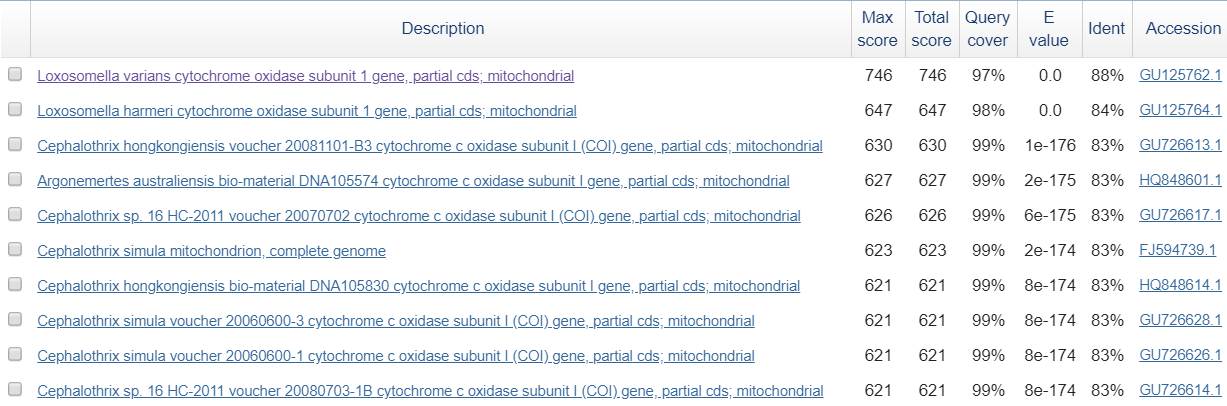

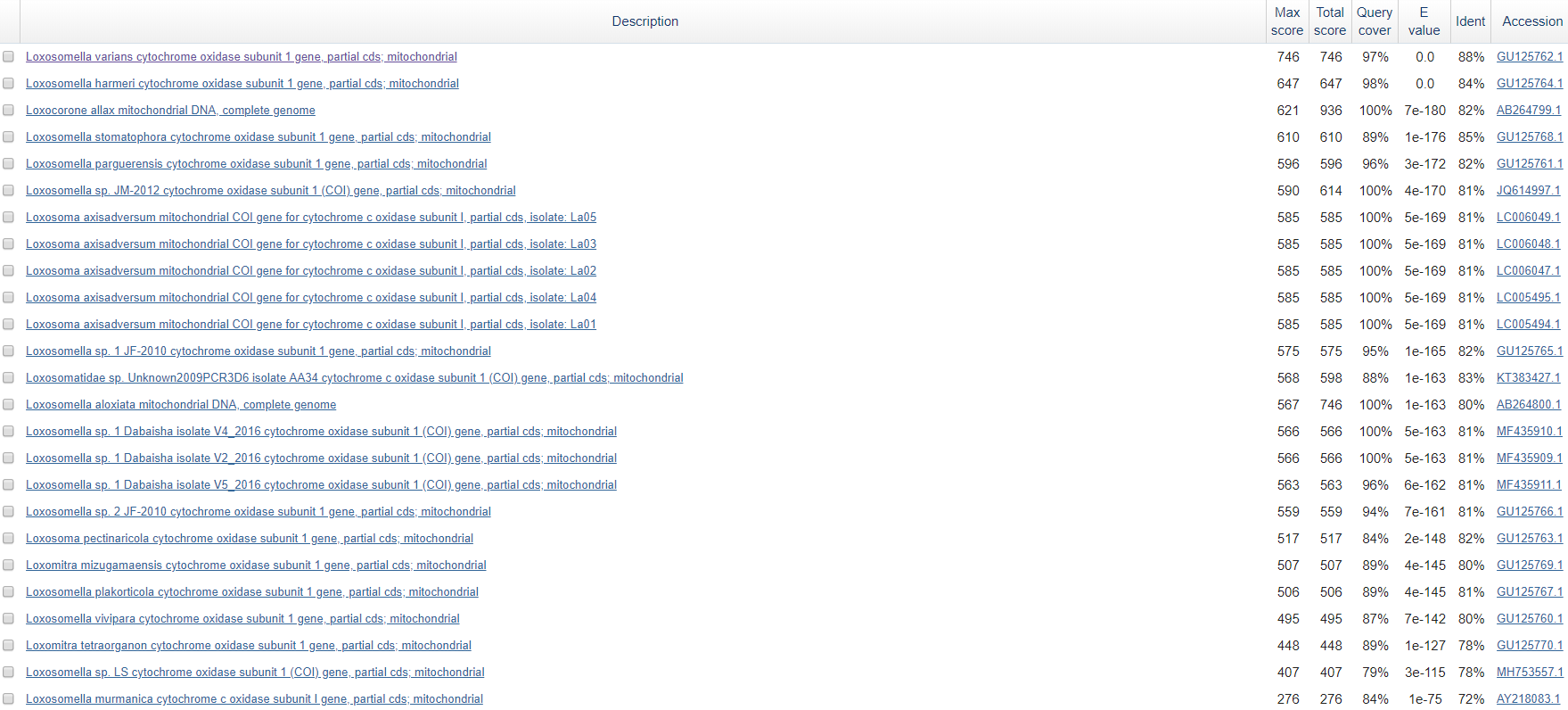
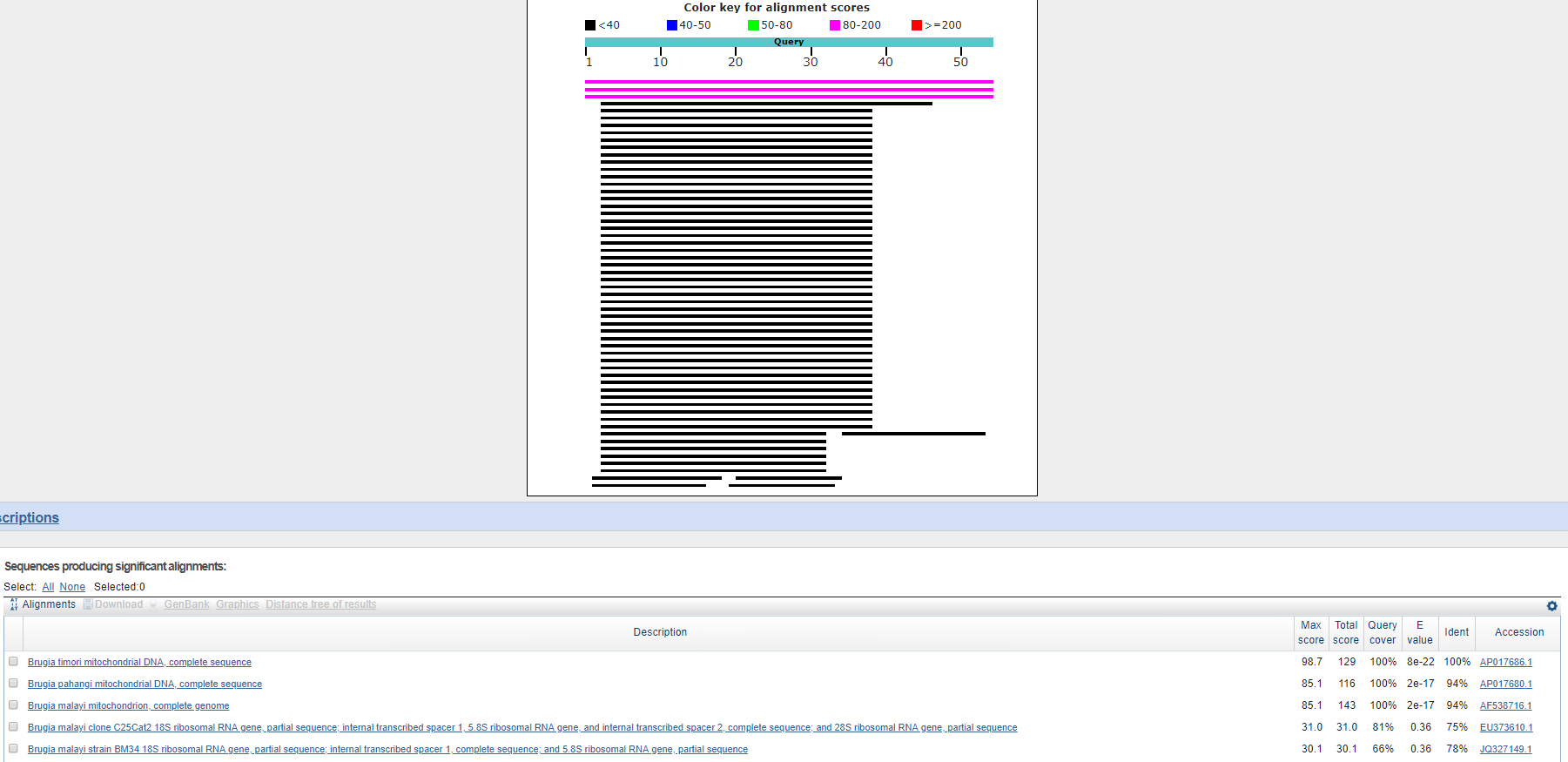
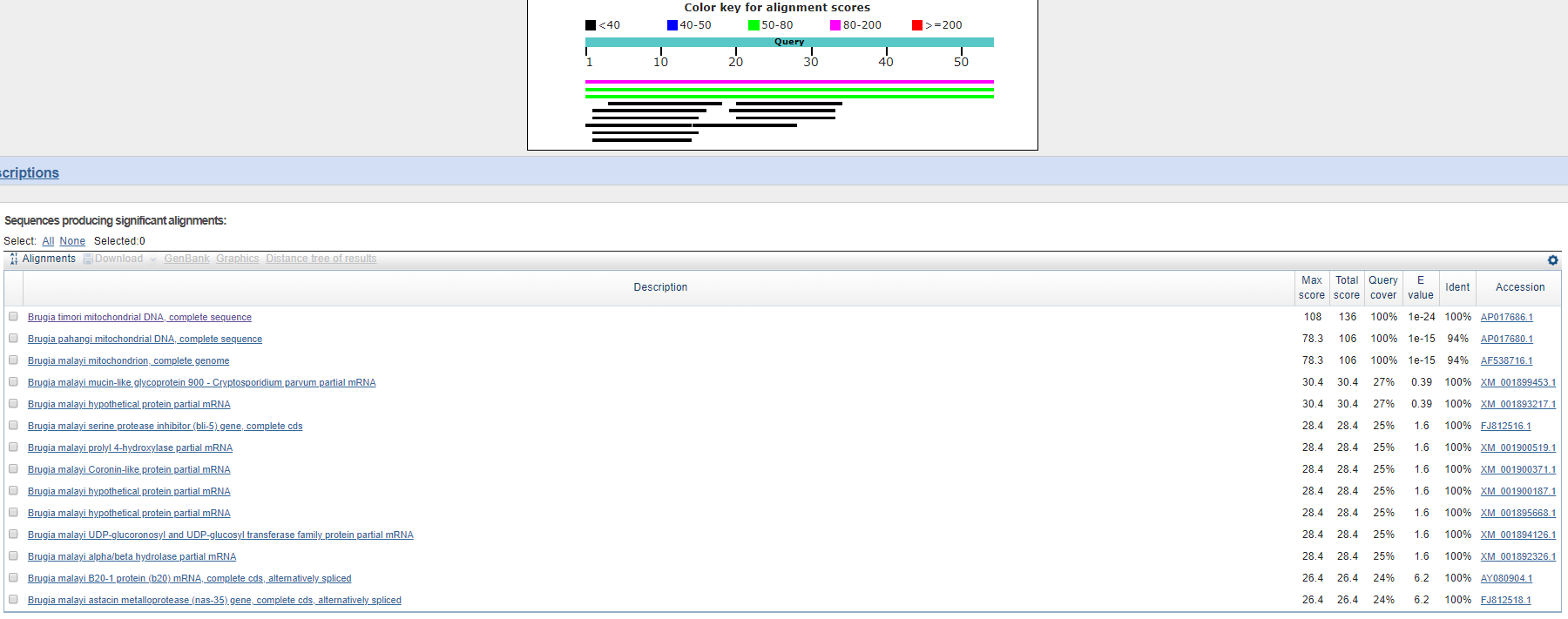
Можно сказать, что нуклеотидная последовательность относится к гену, кодирующему субъединицу I цитохромоксидазы у организма рода Loxosomella, относящегося к семейству Loxosomatidae типа Entoprocta.
| Алгоритм | Параметры алгоритма | Число находок | Комментарии |
| megablast | Стандартные; длина слова = 28; M/M Score 1,-2; |
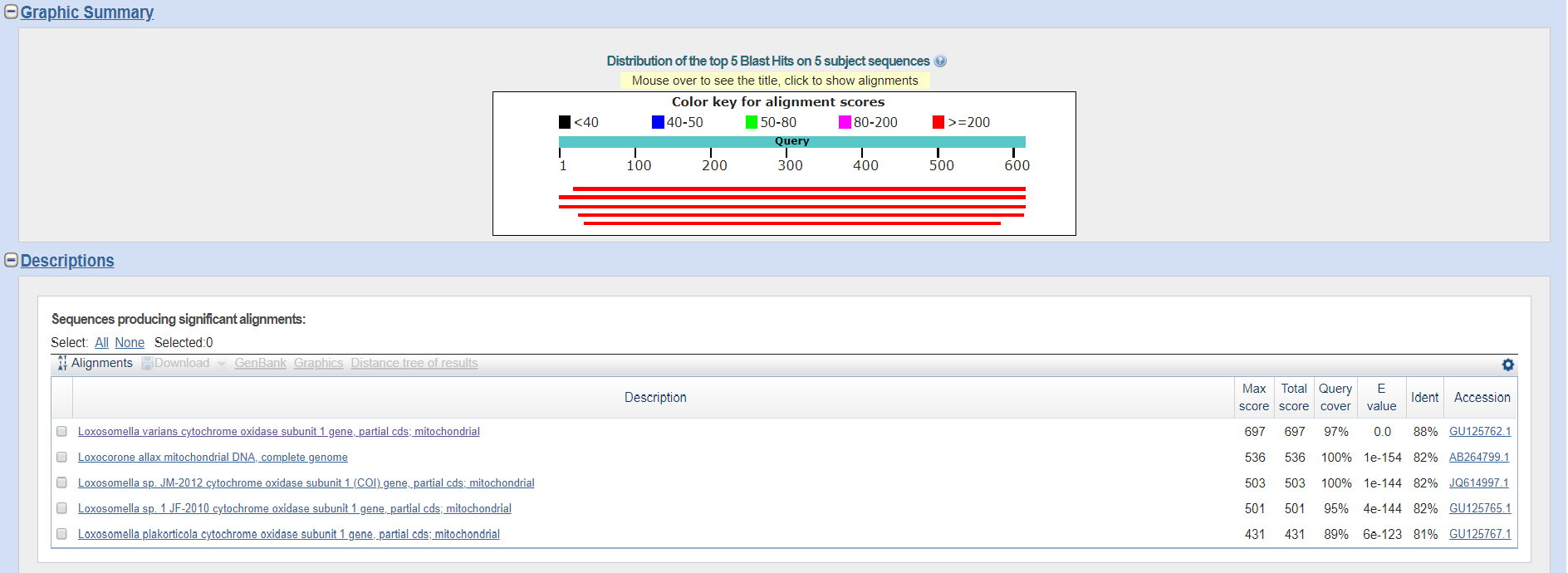
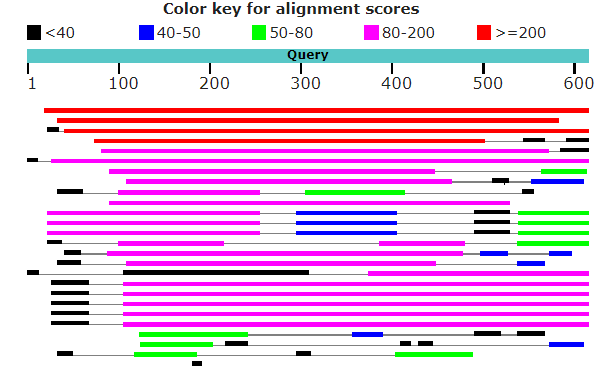
5 | Последняя находка с E-value = 6e-123; первая = 0.0; поиск ограничен семейством; минимальный Ident = 81%; |
| blastn default | Стандартные; длина слова = 11; M/M Score 2,-3; |
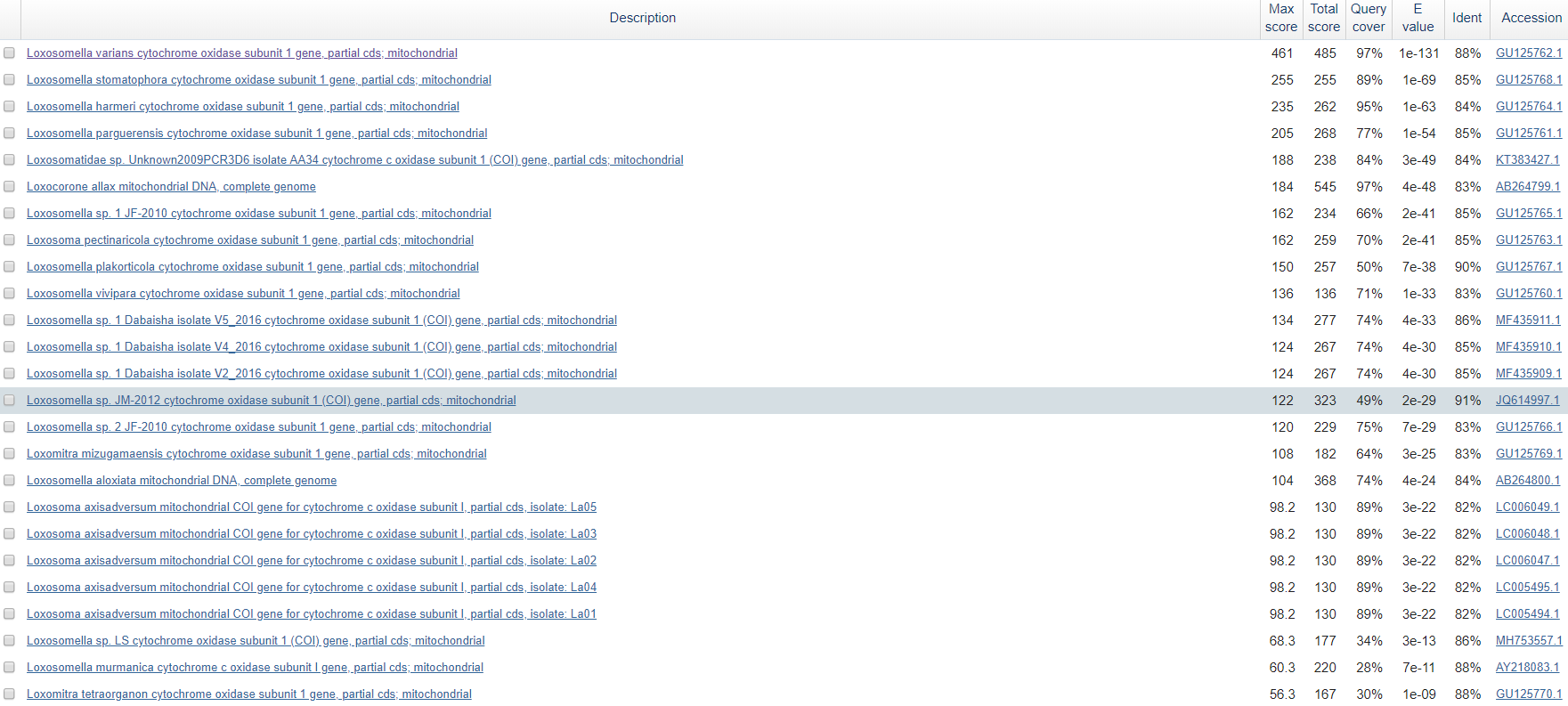
25 | Поиск ограничен семейством; последняя находка на странице с Е-value = e-75; минимальный Ident = 72%; |
| blastn sensitive | Длина слова = 7; M/M Score 1,-4; | 26 | Поиск ограничен семейством; последняя находка на странице с Е-value = e-9; минимальный Ident = 82%; |


| TERT_SCHPO | PRPC_EMENI | |
| Кодируемый белок | trt1 | mcsA |
| Функция | Теломераза представляет собой рибонуклеопротеиновый фермент, необходимый для репликации хромосомных концов у большинства эукариот. Он удлиняет теломеры. Это обратная транскриптаза, которая добавляет простые повторения последовательности к концам хромосом путем копирования последовательности шаблонов в РНК-компоненте фермента. | Митохондриальный ген. Катализирует синтез (2S, 3S) -2-метилцитрата из пропионил-СоА и оксалоацетата, а также из ацетил-СоА и оксалоацетата с большей эффективностью. Также имеет активность цитратсинтазы и может заменить недостаток активности citА. |
| Лучшее совпадение | scaffold-17, Score: 108, E-value: 1e-23 | scaffold-693, Score: 393, E-value: 6e-121 |
| Результат | Есть короткие идентичные участки без гэпов. Чистой гомологии нет, тем более, что низкий Identity (25%) и Positives (47%). | Находка условно положительная. E-value достаточно низкий, чтобы утверждать гомологию двух белков, но вес недостаточно большой, чтобы однозначно утверждать об одинаковой функции. Есть реально гомологичные участки длинной около 25 а.о. |