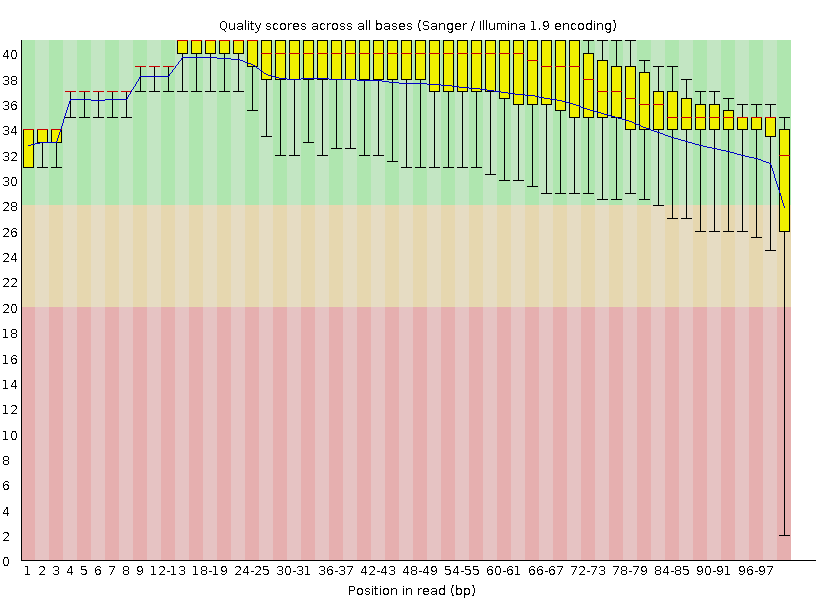
Контроль качества чтений до чистки |
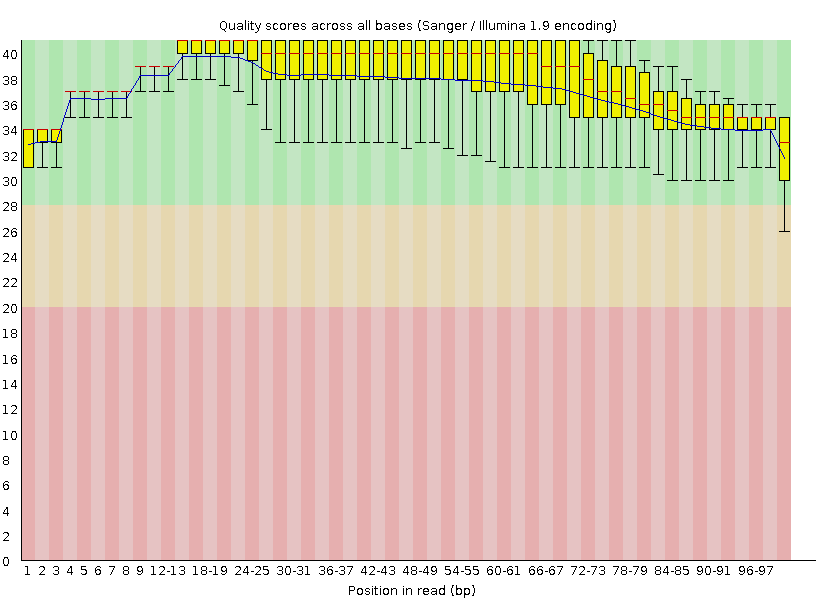
Контроль качества чтений после чистки |
|
Контроль качества чтений до чистки |
Контроль качества чтений после чистки |
| Команда | Функция | Выдача |
| hisat2-build chr10.fasta chr10 | Индексирование референсной последовательности |
Индексированный файл chr10.fasta |
| hisat2 -x chr10 -U chr10_out.fastq --no-spliced-alignment --no-softclip >align1.sam | Выравнивание чтений после чистки с референсной последовательностью | Файл, который содержит выравнивание формата SAM align1.sam |
| Команда | Функция | Выдача |
| samtools view align1.sam -bo align1.bam | Программа переводит файл в формат bam | align1.bam |
| samtools sort align1.bam -T sorted.txt -o sorted.bam | Сортировка выравнивания чтений и референса по координате в референсе | sorted.bam |
| samtools index sorted.bam | Индексирование отсортированного выравнивания | sorted.bam |
| samtools idxstats sorted.bam > totalread.txt | Запись числа откартировавшихся чтений | totalread.txt |
| Команда | Функция | Выдача |
| samtools mpileup -uf chr10.fasta sorted.bam > snp.bcf | Создание файла с полиморфизмами | snp.bcf |
| bcftools call -cv snp.bcf -o snp.vcf | Определения различий | snp.vcf |
| Координата | Тип полиморфизма | В референсе | В чтениях | Покрытие в этом месте | Качество покрытия |
| chr10:5781628 | замена | T | G | 21 | 117.008 |
| chr10:5766152 | индель | AGTAT | AGTATGTAT | 92.4668 | |
| chr10:5766337 | замена | G | A | 93 | 225.009 |
convert2annovar.pl -format vcf4 snp.vcf > chr10.avinput |
Переводим файл .vcf формат, удобный для работы annovar |
annotate_variation.pl -filter -out SR_SNP -build hg19 -dbtype snp138 chr10.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация по Dbsnp |
annotate_variation.pl -out refgen -build hg19 chr10.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация по Refgene |
annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out 1000Genomes chr10.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация по 1000 Genomes |
annotate_variation.pl -regionanno -build hg19 -out GWAS -dbtype gwasCatalog chr10.avinput /nfs/srv/databases/annovar/humandb.old/ |
Аннотация по Gwas |
annotate_variation.pl chr10.avinput -filter -dbtype clinvar_20150629 -buildver hg19 -out CLINVAR /nfs/srv/databases/annovar/humandb.old/ |
Аннотация по Clinvar |
FAM208B 27
RTKN2 28 (+3)
CASP7 6
Помимимо всего прочего в файлах есть информация о синониминости (несинонимичности) замен Остеосаркома 5804531 5804531 G A 70 Ревматоидный артрит 63958112 63958112 T C 75 Vitiligo 115481018 115481018 C T 91ClinVar объединяет информацию о геномных вариациях (полиморфизмах), их отношении к здоровью человека.