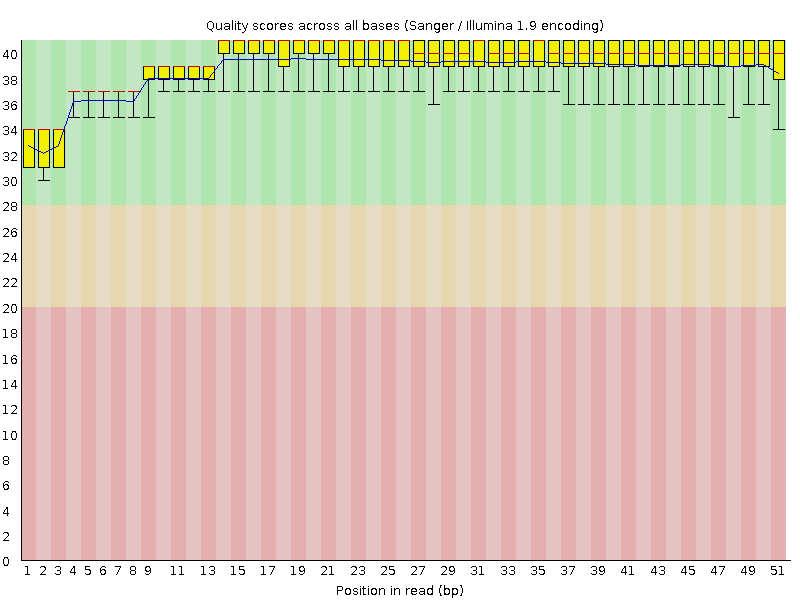
Контроль качества чтений |
|
Контроль качества чтений |
| Команда | Функция | Выдача |
| hisat2-build chr10.fasta chr10.1 | Индексирование референсной последовательности |
Индексированный файл chr10.1.fasta |
| hisat2 -x chr10.1 -U chr10.1_out.fastq --no-softclip >chr10.1.sam | Выравнивание чтений после чистки с референсной последовательностью | Файл, который содержит выравнивание формата SAM chr10.1.sam |
| Команда | Функция | Выдача |
| samtools view chr10.1.sam -bo chr10.1.bam | Программа переводит файл в формат bam | chr10.1.bam |
| samtools sort chr10.1.bam -T nexact.txt -o chr10.1_sort.bam | Сортировка выравнивания чтений и референса по координате в референсе | chr10.1_sort.bam |
| samtools index chr10.1_sort.bam | Индексирование отсортированного выравнивания | chr10.1_sort.bam |
| samtools idxstats chr10.1_sort.bam > result.1.txt | Запись числа откартировавшихся чтений | result.1.txt |
ENSG00000165732.8 14501 ENSG00000266122.1 3 __no_feature 604 __not_aligned 205Опции: