Практикум 12: BLAST
Конспект - запуск BLAST
- Enter Query Sequence
соответственно тут вводится либо ID поледовательности, либо сама последовательность
в поле Query subrange можно ввести диапозон (от и до чего) с которым мы выравниваем
- Choose Search Set
Тут необходимо выбрать базу данных в которой ищем соответствия. По заданию у нас swissprot.
Также можно выбрать в каких организмах (не) искать, и можно исключить несколько классов
послеловательностей
- Program Selection
Выбор нужного алгоритма - зависит от целей.
Интересен DELTA-BLAST (Domain) - который быстро находит схожие домены
- General Parameters
Max target queries -сколько найденных схожих последовательностей вывести на экран
Short queries - подогнать длину липкого слова если введенная
последовательность слишком короткая.
(то есть уменьшить липкое слово, чтобы сделать поиск более аккуратным, что необходимо если
исследуемая последовательность очень короткая)
Expect threshold - оно же E-value. Примерно - математическое ожидание случайно получить
данное выравнивание. Прямо зависит от размера БД и длины последовательности, обратно от
схожести двух последовательностей. Таким образом чем меньше Е, тем более значимо сходство.
В этом параметре можно задать порог значимости выводимых последовательностей.
Соответственно чем меньшее число мы укажем
(формат 1e-9), тем больше последовательностей программа выкинет.
Word size - насколько я поняла, BLAST сначала находит точные (или очень близкие
в случае аминокислот) соответствия коротких слов (назовем их липкими)
между двумя последовательностями,
а потом уже дальше смотрит на совпадения. Таким образом, уменьшив длину липкого слова, мы
увеличим тщательность поиска (и удлинним время работы)
Max matches in a query range
- Scoring Parameters
Matrix - матрица замен, по которой вычисляется вес (то есть +-сходство) двух последовательностей.
(если у нас аминокислоты, то замена валина на лейцин в ходе эволюции гомологичных
белков более вероятна, чем замена глицина на триптофан)
Gap costs - опять же параметр для вычисления веса выравнивания - насколько "дорого" (то есть вероятно)
образования гэпа в ходе эволюции. Соответственно если мы этот параметр увеличим
то алгоритм будет выдавать выравнивания где будет меньше гэпов. А когда и до каких пор мы имеем право это
делать - вопрос к тому как последовательности эволюционируют.
Compositional adjustments - как было сказано в презентации, "борьба с участками малой сложности".
То есть насколько я понимаю, (!) программа сама улавливает напрмер перепредставленность
какой-нибудь аминокислоты во введенной последовательности и в соответствии с этим изменяет
значения в матрице сходства.
Задание 1. Гомологичные последовательности
Ссылка на отчетную табличку
Ссылка на Jalview проект

В принципе, по приведенным параметрам эти тоже можно считать гомологичными.
Удалила я из первоначального набора только Probable 2-carboxy-D-arabinitol-1-phosphatase (как ни странно,
Е = 1е-19) и 2,3-bisphosphoglycerate-dependent phosphoglycerate mutase (Е = 8е-07). Осталась
и последовательность с Е = 0.004 (она самая нижняя).
Обоснуем гомологичность: Участок с 55 по 93 начинается и заканчивается 100% консервативными позициями,
длиной он 38 аминокислот, колонок с гэпами там нет (кроме оставшейся от множественного выравнивания)
Плотность консервативных позиций не очень видна на пороге above 90 для 11 последовательностей,
но ставим 50% и получаем другую картинку:
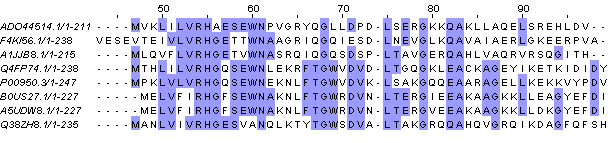
Почему так вышло? Замечу, что на самом деле выявленные BLAST белки не сильно варьировались по
названиям, и почти везде было 'phosphotase'. Плюс весь низ таблицы занимали белки
2,3-bisphosphoglycerate-dependent phosphoglycerate mutase, что и из выравнивания видно (нижние белки
между собой обладают очень большим сходством).
Можно сделать вывод, что при заданных параметрах поиска (все брались исходные),
бласт действительно выдает в большинстве своем белки гомологичные.
Видимо, фосфатаза и мутаза имели общего предка - функции хоть и немного различны,
но субстрат и катализируемая реакция, а значит и конформация и строение не должны сильно различаться.
Задание 2. Карта сходства
Взяты A0A163IMY9_DIDRA и A0A0M2LYI0_9MICO.
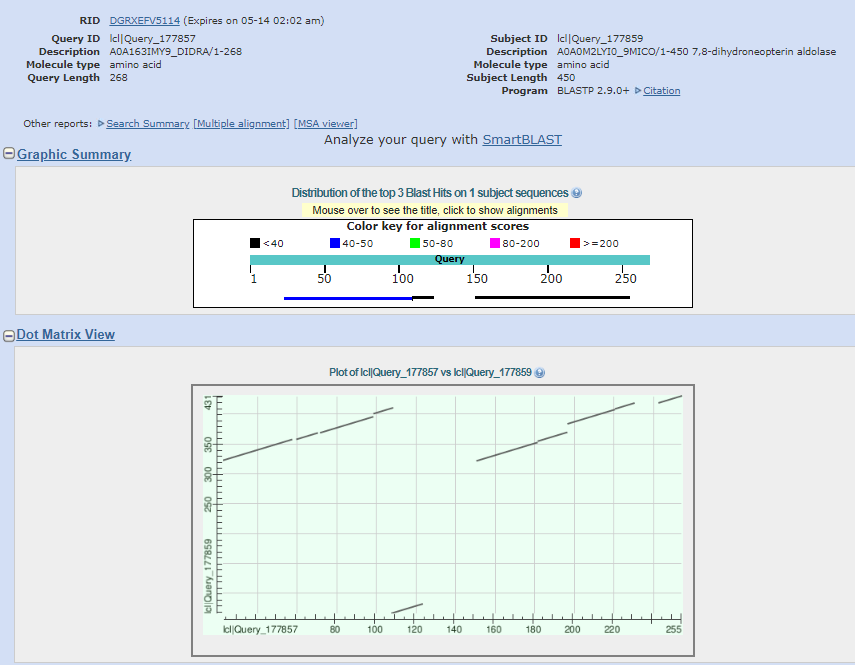
Если я правильно понимаю, вторая последовательность (та что выставлена по горизонтали - DIDRA)
является дважды повторенным куском конца последовательности MICO. То есть это дупликация
второго участка. Тк наклон положительный, то переворачиваний не происходило. Разрывы со
смещением соответствуют вставке каких-либо аминокислот.
Задание 3. Игры с BLAST
Некодирующая последовательность - На что стоит обратить внимание
Написалась программка, выдающая случайную аминокислотную последовательность:
import random
length = int(input())
s = str()
for i in range(length):
s += random.choice('GAVLICMFYWPSTNQDEHKR')
print(s)
Сначала на гипотетическом белке длиной 200 аминокислот
Запуск с обычными параметрами (я не уследила и БД - nr) дал 4 последовательности, причем минимальное
E было 0.21
С wordcount 2 выдача дала уже 6 последовательностей, Е снова в дипозоне от 0.21 до 10(то что до 10
не удивительно оно в Expected по умолчанию стоит)
Wordcount 2 и Gap Costs снижен до 9 (extention все еще 1). Белков в выдаче 5, из них 2 совсем новых.
Тот с которым Е было 0.21 теперь 0.73. Я снизила штраф за индели, что в целом упростило выравнивание
с моей последовательностью, а значит матожидание выросло.
В общем можно сделать вывод, что совсем рандомную последовательность большой длины - штука редкая,
и не будет особо ни с чем выравниваться. На самом деле моя програмка исходит из того,
что все аминокислоты встречаются в природе с одинаковой вероятностью, что, конечно неправда
и учитывается при построении выравниваний (для этого и есть матрицы сходства)
А теперь возьмем белок длиной 30 аминокислот
Я взяла wordcount 2 и галочка на adjustment. Снова минимальное E 0.33, количество последовательностей
стало более 20000.
Видимо отличия от рабочих пептидов замтны даже на 30 аминокислотах!
Мой белок - wordsize
БД - swissprot
Выдача составила (в скобочках количество выдач с Е меньше 1e-15):
2 - 422 (15)
3 - 421 (15)
6 - 344 (14)
В целом конечно заметно более вкрадчивый поиск - схожих белков обнаружилось больше
Коллаген и Compositional adjustments
Я взяла коллаген гидры (а в коллагене перепредставлены G и P ) - BAA23625.1
И запустила с wordcount 6.
для conditional compositional score matrix adjustment выдача составила 257 белков
а для no adjustment - 500 белков
причем E во втором случае были в целом выше
Вывод: лучше там ставить галочку, подстраивание матрицы это полезно!