Хромосома № 8
| cp Human/chr8.fasta sofyagdk26/chr8.fasta | Копирование необходимых файлов в свою директорию |
|---|---|
| hisat2-build chr8.fasta indexed | Индексация референса |
| cp ../Human/reads/chr8.fastq chr8.fastq | Копирование |
| fastqc chr8.fastq | Анализ программой FastQC. Потом смотреть на созданный html файл |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr8.fastq chr8_trimmed.fastq TRAILING:20 MINLEN:50 | Очистка при помощи Trimmomatic. Адаптеры уже были удалены. Чтения одноконцевые! Необходимо только обрезать с конца нуклеотды с качество ниже 20, и убрать все последовательности длиной менее 50 нк |
| hisat2 -x indexed -U chr8_trimmed.fastq -S chr8_aligntoref.sam --no-softclip --no-spliced-alignment | Картирование чтений из fastq файла на проиндексированную последовательность; --nosoftclip - выравнивание должно быть по всему риду; --no-spliced-alignment - выравнивание не должно допускать гэпов и инделей в риде |
| samtools view -b chr8_aligntoref.sam -o chr8_aligntoref.bam | Перевод в бинарный bam формат |
| samtools sort chr8_aligntoref.bam chr8_altoref_sorted | Сортировка выравниваний по координате в референсе |
| samtools index chr8_altoref_sorted.bam | Индексирование |
| samtools mpileup -u -f chr8.fasta -o chr8_polymorph.bcf chr8_altoref_sorted.bam | Создание файла с полиморфизмами |
| bcftools call -cv -o chr8_polymorph.vcf chr8_polymorph.bcf | Изменение формата, -v - output variant sites only - то есть показать только изменившиеся позиции |
| vcftools --vcf chr8_polymorph.vcf --remove-indels --recode --out chr8_polymorph_noindel | Удалить из vcf файла индели (вывод программы: After filtering, kept 95 out of a possible 100 Sites) |
| convert2annovar.pl -format vcf4 chr8_polymorph_noindel.recode.vcf -outfile chr8_polymorph.avinput | Конвертация в формат, который распознает annovar |
| annotate_variation.pl -out ann_chr8_refgene -build hg19 -dbtype refGene chr8_polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотация по refgene, на генной разметке |
| annotate_variation.pl -filter -out ann_chr8_dbsnp -build hg19 -dbtype snp138 chr8_polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотацтия по dbsnp, основана на фильтрации |
| annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out ann_chr8_1000g chr8_polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотацтия по 1000 genomes, основана на фильтрации |
| annotate_variation.pl -regionanno -build hg19 -out ann_chr8_gwas -dbtype gwasCatalog chr8_polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотацтия по GWAS, основана на разметке других регионов генома |
| annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 -out ann_chr8_clinvar chr8_polymorph.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотацтия по Clinvar, основана на фильтрации |
Изначально было 8367 чтений.
Результат работы Trimmomatic:
Input Reads: 8367 Surviving: 8227 (98,33%) Dropped: 140 (1,67%)До обрезки
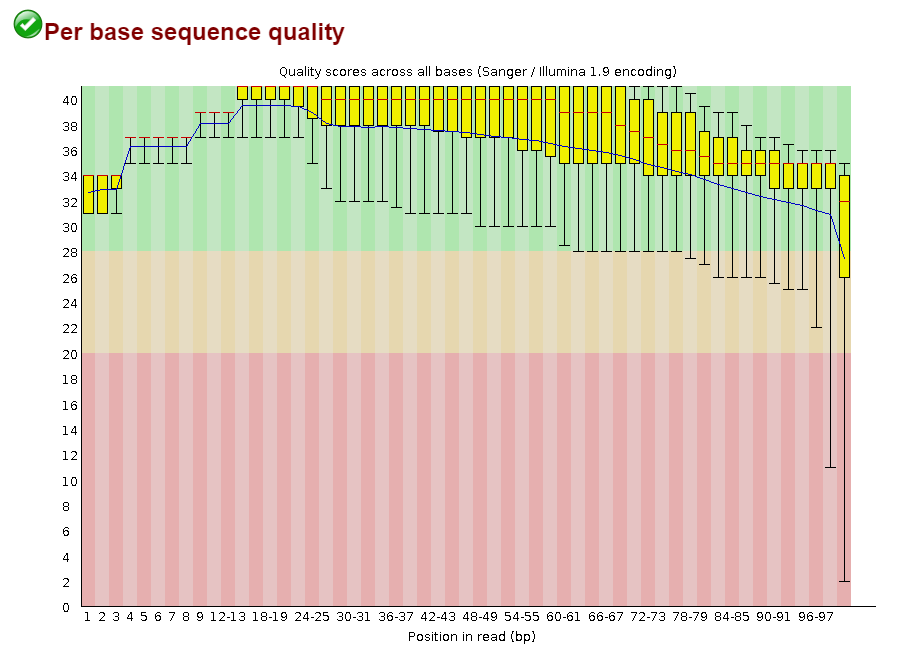
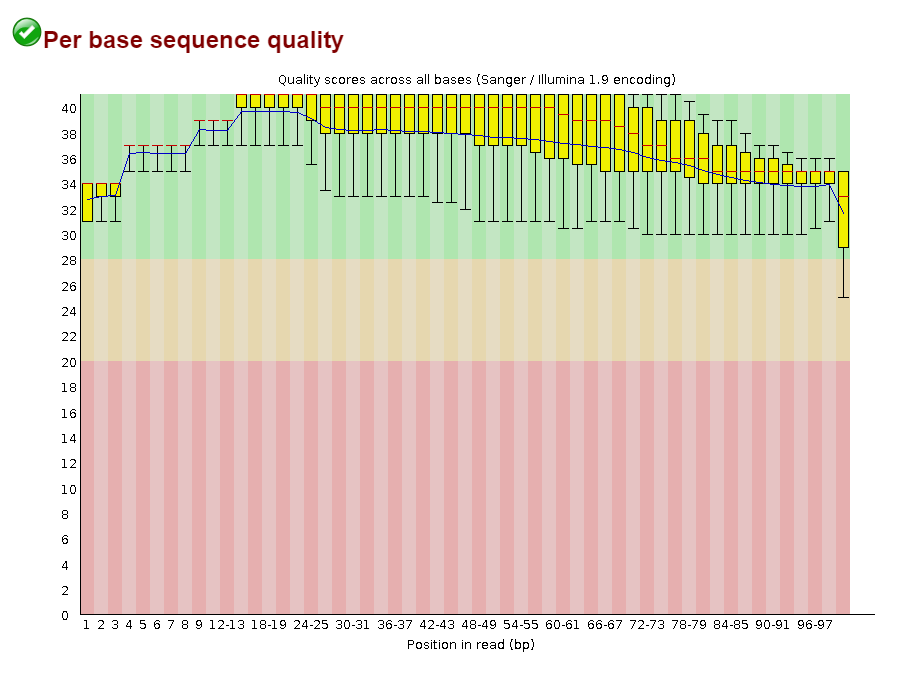
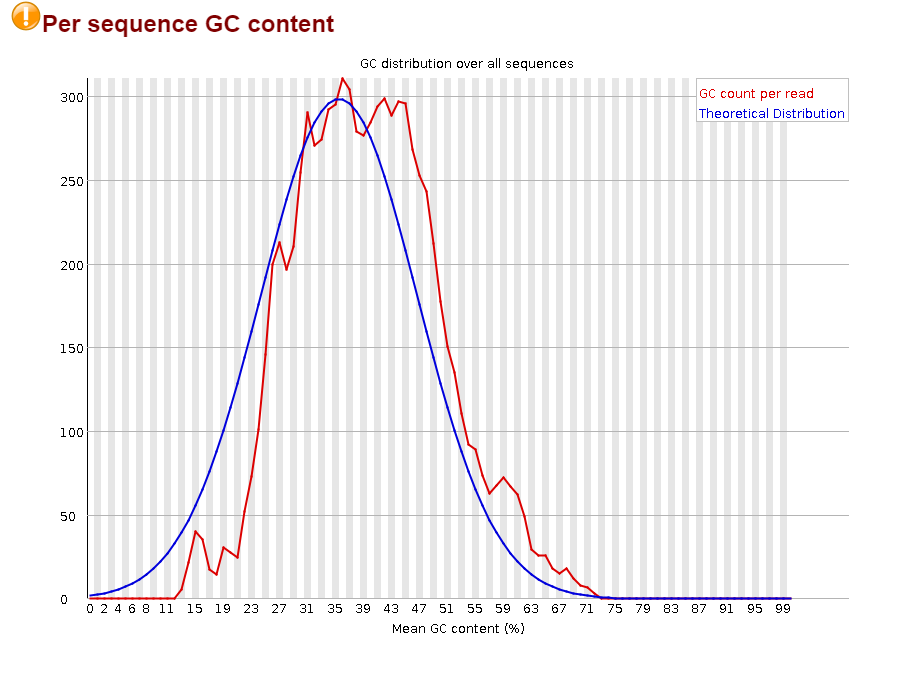
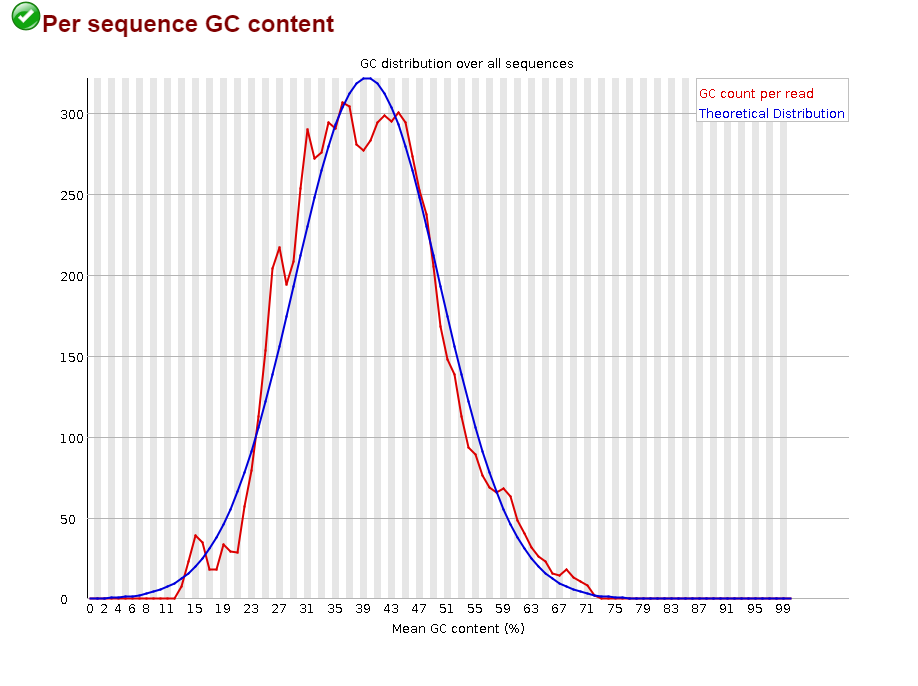
Откартировано 99.64% чтений
Вывод программы hisat2:
8227 reads; of these:
8227 (100.00%) were unpaired; of these:
30 (0.36%) aligned 0 times
8197 (99.64%) aligned exactly 1 time
0 (0.00%) aligned >1 times
99.64% overall alignment rate
Вывод: Кажется, это очень хорошее качество картирования!
Насколько я поняла из
документации по формату, страницы 4-5 , данные о глубине покрытия можно изкать в колонке INFO после
ключа DP.
| строка в файле /nfs/srv/databases/ngs/sofyagdk26/chr8_polymorph.vcf | тип полиморфизма | референс | чтения | глубина покрытия | качество чтения |
|---|---|---|---|---|---|
| 39 | замена | С | Т | 17 | 111.008 |
| 75 | делеция | GTTTTTTTTTTTT | GTTTTTTTTTT,GTTTTTTTTT | 74 | 120.457 |
| 90 | вставка | А | АС | 28 | 57.456 |
У меня получилось в итоге 95 SNPs (64 transitions and 31 transversions) и 5 инделей
Качество
Медиана - 11.3429
Среднее - 66.056
Глубина
медиана - 2
среднее - 14.57
| категория | количество SNP в группе |
| UTR3 | 13 |
| intronic | 60 |
| exonic | 5 |
| intergenic | 17 |
| строка в файле | ген | к каким заменам привело |
|---|---|---|
| 5 | CLU:exon5 | synonymous A G |
| 28 | HNF4G:exon1 | nonsynonymous G A |
| 38 | HNF4G:exon8 | synonymous G A |
| 39 | HNF4G:exon8 | nonsynonymous G A |
| 77 | TRPS1:exon2 | synonymous C A |
RS имеет 77 SNP из 95 - по результатам аннотирования относительно dnsnp
Средняя частота 0.463293886 - по результатам выравнивания относительно бд 1000genomes
Меня очень удивила такая вещь - я посмотрела частоту замен для
предсказанных экзонных snp
и вышло так: для синонимичных замен частоты были 0.66, 0.00499, 0.16853; а для несинонимичных -
0.61 и 0.62. Common sense мне подсказывал, что по крайней мере частота синонимичных замен
должна быть обычно достаточно высокой - против них же не идет прямого отбора. Но может
тут выборка и common sence просто подводят.
В аннотации Clinvar ничего не было найдено - файл пуст! Насколько я понимаю, тут нужно смотреть в выдачу аннотации GWAS
gwasCatalog Name=Alzheimer's disease chr8 27456253 27456253 T C het 31.0117 . gwasCatalog Name=Alzheimer's disease (late onset) chr8 27466315 27466315 T C hom 154.998 . gwasCatalog Name=Urate levels chr8 76478768 76478768 C T hom 221.999 . gwasCatalog Name=HDL cholesterol chr8 116599199 116599199 T G hom 221.999 .Интересно соотнести с выдачей refseq: оказалось, что первые 2 - в итнтронах гена CLU, вторая находится в UTR3 гена HNF4G, a 4ая в интроне гена TRPS1. Как оказалось ген CLU кодирует белок кластерин - белок теплового шока, молекулярный шаперон, работающий в аппарате Гольджи и регулирующий сворачивание секретируемых белков. Его недопроизводство может служить импульсом для синтеза p53 - белка, участвующего в апоптозе. Понимая болезнь Альцгеймера на самом простом уровне как дегенеративое заболевание определнных тканей головного мозга действительно можно предположить, что ткань ГМ будет чувствительная к изменениям в механизмах апоптоза клеток в ответ на изменения в вырезании интронов в белке CLU.