Качество предоставленных прочтений необходимо было проверить с помощью программы FastQC, предустановленной на kodomo.
На Рисунке 1А изображена выдача программы.
Результатом работы этой программы стал архив chr14_fastqc.zip и илюстративный 'chr14_fastq.html'? кторый визуализирует некоторую информацию о чтениях.
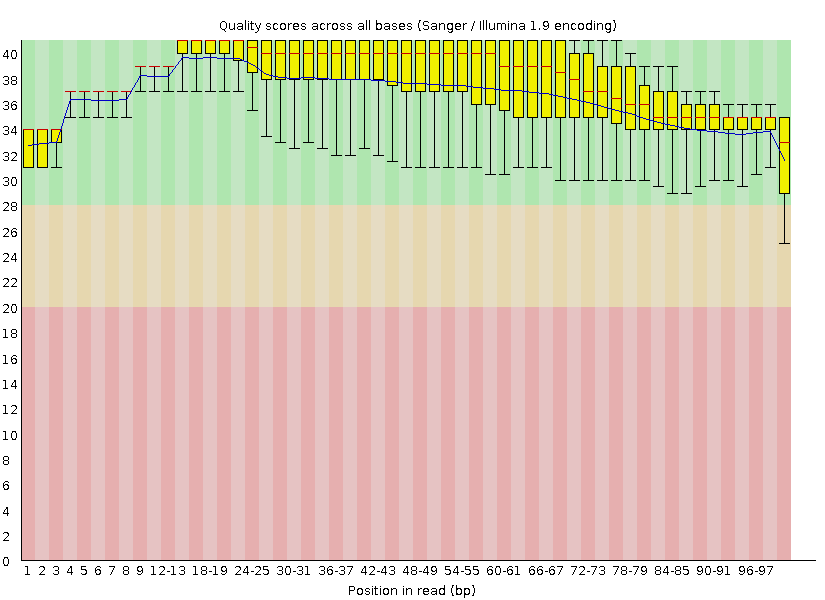
Ниже представлена илюстрация из html-выдачи: "Per base quality", на которой изображено качество определения конкретного нуклеотида на каждой позиции рида.
Поле гистограммы поделено на три полосы, по оси ордтнат откладывается значение качества, так что
интутитивно и илюстративно понятно, что чем выше качество, тем лучше прочтение - зелёная зона является предпочтительной.
Для анализа использованы такие статичтические характеристики, как:
- Среднее значение качества (синяя линия)
- Медиана значений (красные линии на интервале)
- Интервальный размах (жёлтый прямоугольник) - диапазон, в котором четверть прочтений имеет значение качества по данной позиции не выше нижней границы, а три четверти - не выше верхней.
- Интервальный размах между десятым и девяностым процентилем (черная риска погрешности) - то же самое, что и выше, но с 10% и 90% процентами чтений
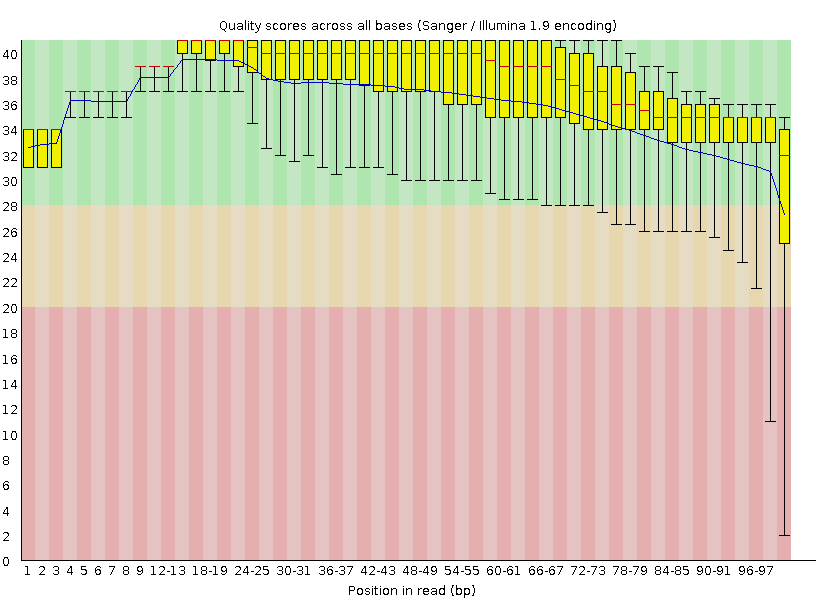
Конец ридов закономерно прочитан хуже, западание на область "среднего качества" наблюдается уже 73-й позиции, с 60-72 позицию значения близки к нежелательному диапазону - то есть около 40% позиций вызывают сомнение. Это обьясняется тем, что секвенирование копит ошибки, и с увеличением длины риды процентное содержание проблемных мест в нём растёт. Именно поэтому риды не представлены, по-существу, очень длинным фрагментами - те не репрезентативны.
Очистка осуществлялась с помощью программы Trimmomatic.

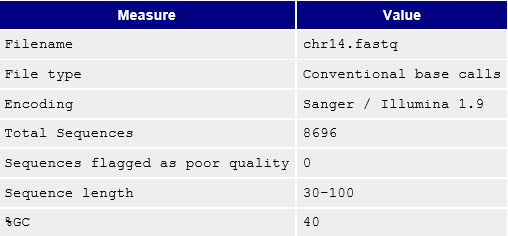
На вход команда приняла 8696 ридов, оставив только 8562 - 98.46% от общего количества. Были удалены концы каждго чтения с качеством ниже двадцати по вышеозначенному графику, и все короче 50 пар нуклотидов.
| ДО: | ПОСЛЕ: |
 |
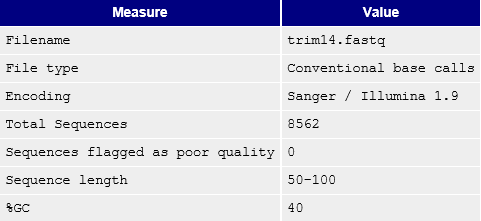 |
| Рисунок 1С Basic statistic. Сравнение. | |
|
 |
| Рисунок 1D Per base quality. Сравнение. | |
На Рисунке 1С и 1D представлены два пункта выдачи FastQC, до и после чистки. Теперь наглядно можно наблюдать, какие риды были удалены Trimmomatic. Среднее значение сильно поднялось к концу прочтения, ныне лишь последний фрагмент заходит в зону среднего качества, чего, видимо, было не избежать, потому как ранее последний диапазон заходи в красную, совершенно непригодную зону. Изменились длина и число ридов. Рисунок 1С предоставляет характеристики к сравнению: теперь ддины последовательностей варьирутся от 50 до 100 в противовес нижней границе в 30 пар оснований до чистки.
Второй этап: картирование чтений.
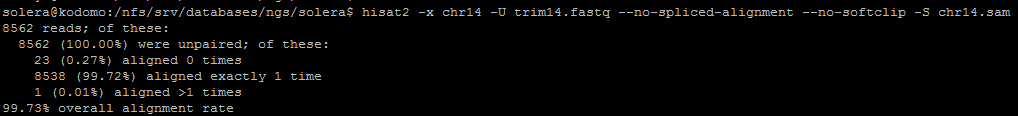
С помощью программы Hisat2 были откартированы очищенные чтения.
Необходимые данные для запуска были перемещены в рабочую директорию и проведён запуск программы:
Слишком обьёмный для иллюстраии log программы доступен к просмотру в файле loghisat.txt,
выдача - в файле hisat.txt.
Программа сконструировала восемь файлов вида chr14.*.ht2, где * - цифра от 1 до 8.
После того, как референсная последовательность была проиндексирована, очищенные чтения были выровнены по индексированной последовательности
- файлам выдачи без разрывов(параметр -no splieced alignment) и подрезания ридов (-no softclip), типичного для концевых участков.
С помощью пакета samtools была произведён анализ выравнивания.
Сначала файл чтений был переведён в формат .bam из формата .sam выдачи hisat (см. предыдущий параграф).
Запуск программы представлен ниже.
- -b -- выходной файл бинарный
- -o -- указание имени выходного файла
Далее происходила сортировка выравнивание чтений с референсом (получившийся после картирования .bam файл) по координате начала чтения. Опция -Т с последующим указанием файла это операция перенаправения временных файлов.

Далее отсортированный файл .bam был проиндексирован, с послежующим выяснением, сколько чтений, в итоге, было откартировано на геном: команды можно наблюдать на Рисунке 3B.
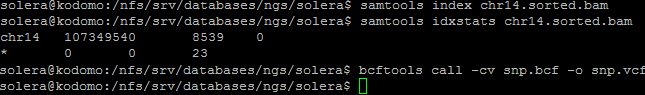
Программа выдала следующий анализ: после названия последовательности идут, последовательно, её длина, число картировавщихся ридов, число некартировавшихся ридов. Можем сравнить данную выдачу с логом программы хисат (Рисунок 2А). Действительно, из 8539 картированных по мнению indxstats ридов все упомянуты hisat, а один, как видно из Рисунка 2А, даже прокартировался более одного раза.
Итого:
- Число невыровненных ридов: 23(0.27%)
- Число ридов, выровненных 1 раз: 8538(99.71%)
- Число ридов, выровненных более 1 раза: 1 (0.01%)
Для анализа так же доступен файл chr14.sam - файл выдачи hisat2.
Третий этап: анализ SNP .
Файл с полиморфизмами был создан с помощью всё той же программы пакета samtools - samtools mpileup.

Ниже представлены три полиморфизма, выбранные из списка полиморфизмов vsp-файла. С полным можно ознакомиться скачав его.
| CHROM | POS | REF | ALT | QUAL | INFO | FORMAT | chr14.sorted.bam |
| chr14 | 81448951 | GAAAAAAAAAA | GAAAAAAAAAAAA, GAAAAAAAAAAA, GAAAAAAAAAAAAA | 79.4672 | INDEL;IDV=40;IMF=0.754717;DP=53;VDB=0.765077; SGB=-0.693146;MQSB=1;MQ0F=0;AF1=1;AC1=2; DP4=3,1,21,21;MQ=60;FQ=-63.5253;PV4=0.609302,1,1,1 | GT:PL | 1/1:153,62,33,147,0,112,148,67,154,141 |
| chr14 | 81467864 | CAT | C | 217.468 | INDEL;IDV=7;IMF=0.4375;DP=16;VDB=0.365321; SGB=-0.636426;MQSB=1;MQ0F=0;AF1=0.5;AC1=1; DP4=5,4,5,2;MQ=60;FQ=217.468;PV4=0.632867,3.98565e-10,1,1 | GT:PL | 0/1:255,0,255 |
| chr14 | 81490813 | G | A | 9.52546 | DP=1;SGB=-0.379885;MQ0F=0;AF1=1;AC1=2; DP4=0,0,0,1;MQ=60;FQ=-29.9906 | GT:PL | 1/1:39,3,0 |
Фрагмент файла выдачи даёт информацию о: позиции полиморфизма(POS), участке в референсной последовательности(REF) и аналогичном участке в чтении(ALT), качестве покрытия(QUAL), информации о типе полиморфизма по мнению программы(INFO), данные о генотипе: 0/1 - диплоидный, гетерозигота (1 - аллель в выравнивании, 0 - в референсе), вероятности этих генотипов=PL(FORMAT и sorted.bam). Но мы позволим себе интерпретировать полиморфизмы по-своему.
- Первый: двукратная дуплекация участка относительно референсной последовательности. Качество, относительно других прочтений, низкое, но не слишком - самое большое значение варьировалось в районе 200. Глубина покрытия (DP) - 53.
- Второй: вставка. Качество очень высокое, в десятке лучших. Глубина покрытия - 16.
- Третий: замена. Качество очень низкое, к сожалению, всего 9, глубина покрытия - 1.
С помощью программы annovar были проаннтотированы все замены, не включая индели первоначальнрого файла .vcf.
Сначала был сконструирован vcf-файл без инделей, после этот файл был переведён в нужный формат.

- Наличие идентификатора rs проверялось по базее данных dbsnp (лист snp-rs). Оказалось, что у шести snp из всез 88 нет такого идентификатора.
- Частоты замен были определены по базе 1000genomes, полная аннотация на соответствующем листе таблицы (1000genomes). Самыми распространёнными оказались частоты замен от ~0.3 до ~0.5, медиана = 0.23.
- Известные клинические последствия данных замен были аннотированные по базе Clinvar. Выходной файл оказался пуст, так как для данных snp, видимо, нет известных клинических последствий.
- Клиническая аннотация была получена из базы GWAS (лист gwas).
- Из 88 аннотированных по refgene snp 56 имеют гомо-(hom) замену, и оставшиеся 22 имеют гетеро-(het) замену.
Из них 3 попали в экзоны, одна в сплайсинг-вариант, одна в UTR3 нетранслируемую область, одна в междугеновое (intergenic) пространство.
Остальные попали в интроны.
Замены наблюдаются в генах RNASE9, PPP2R5C, TSHR и в локусе LOC101927081,LINC00645 (лист variant_function).
Замены в генах наблюдается три, по одной на экзоны каждого гена (лист exonic_function). Из 3 произошедших замен в экзонах, только замена в гене TSHR является синонимичной, когда как другие замены привели к замене аминокислоты.
Описанные выше гены:
- TSHR - ген рецептора тироид-стимулирующего гормона
- RNASE9 - ген рибонуклеазы 9 из семейства РНКаз А
- PPP2R5C - ген регуляторной субъединицы гамма белковой фосфатазы-2
| Prostate cancer | RNASE9 | 21024619 | exon | A | G |
| Graves'disease | TSHR | 81451229 | intron | T | C |
| Autism | PPP2R5C | 102360745 | intron | G | C |
У пациентов с такими заболеваниями были найдены вышеозначнные мутации, что может, гипотетически, как-то коррелировать с болезнью (ассоциироваться) - а может и нет.