Первый этап: подготовительный
В рабочую директорию /nfs/srv/databases/ngs/solera/pr12
были получены две биологические реплики одноконцевых чтений в формате fastq
из каталога /P/y14/term3/block4/SNP/rnaseq_reads.
Разметка генома в формате .gtf по версии Gencode19 для сборки hg19
была реквестирована по тому же адресу.
Этап второй: анализ качества чтений
Качество предоставленных прочтений необходимо было
проверить с помощью программы
FastQC, предустановленной на kodomo.
На Рисунках 1 и 2 изображены выдачи программы для каждой из реплик.
Ознакомиться с полной html-выдачей так для первой и второй реплик можно, пройдя по ссылкам.
Результатом работы этой программы стали архивы chr14.*_fastqc.zip, ссылки на которые я приводить не буду,
и илюстративные chr14.1_fastq.html и chr14.2_fastq.html, которые визуализируют некоторую информацию о чтениях - ссылки представлеены выше в контексте.
Подробное описание графического представления "Per base quality" смотри в предыдущем практикуме,
остальные графики достаточно очевидны и наглядны, чтобы посвящать разбору способа построения гистограмм время. Скажем лишь несколько слов о приведённых
ниже графических данных чтений: качество нуклеотидов в ридах, качество ячеей иллюмины, и те данные, напротив которых стояли знаки, отличные от галочек(т. е. c отклонение от "нормальных" - или ожидаемых правильнее).
Для сравнения можно ознакомиться с "хорошими" данными на сайте FastQC.
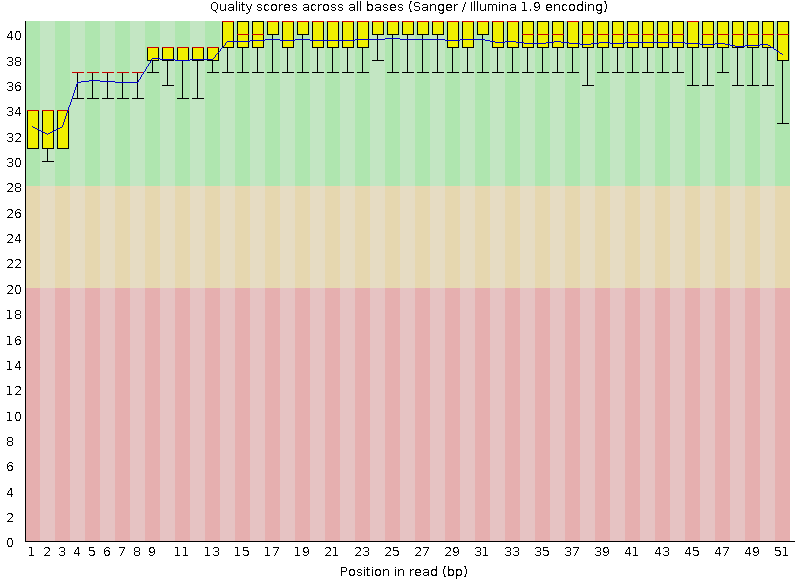 Рисунок 1A. Качество чтения нуклеотидов: первая реплика | 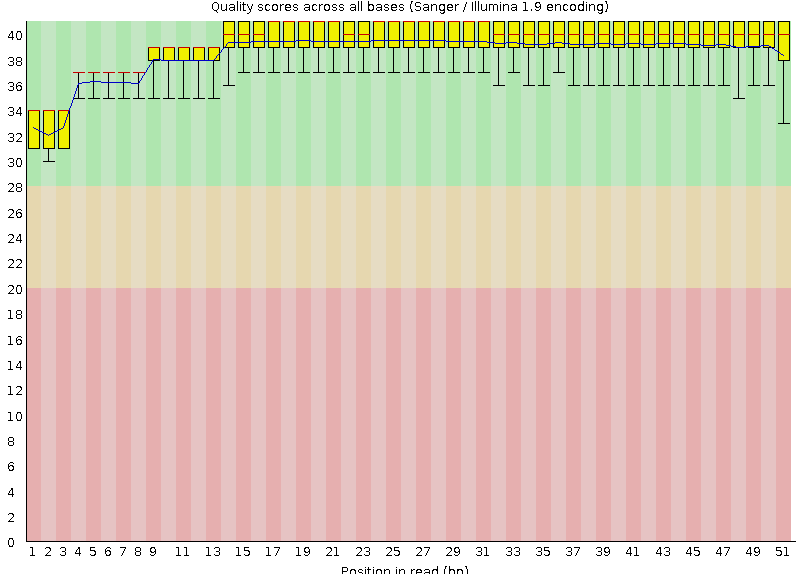 Рисунок 2A. Качество чтения нуклеотидов: вторая реплика |
Оба чтения попдадают в зеленую область даже учитывая децили. | |
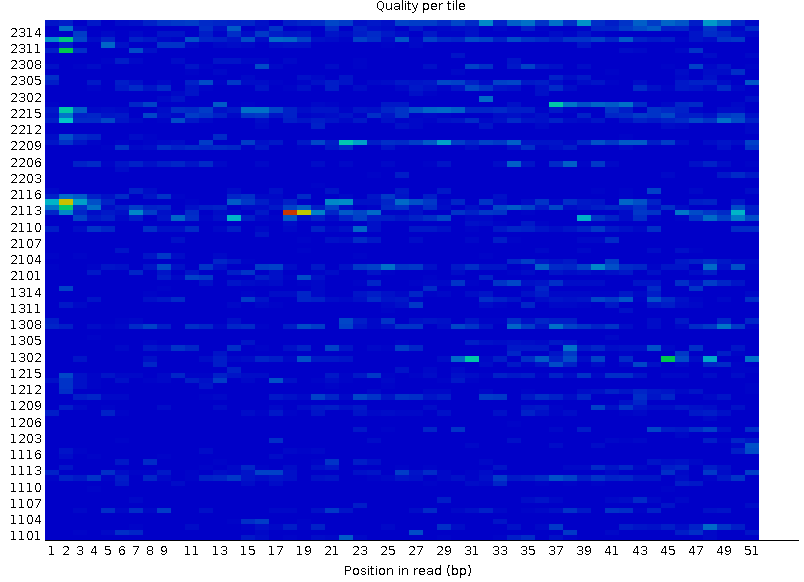 Рисунок 2A. Качество ячеек Иллюмины: первая реплика | 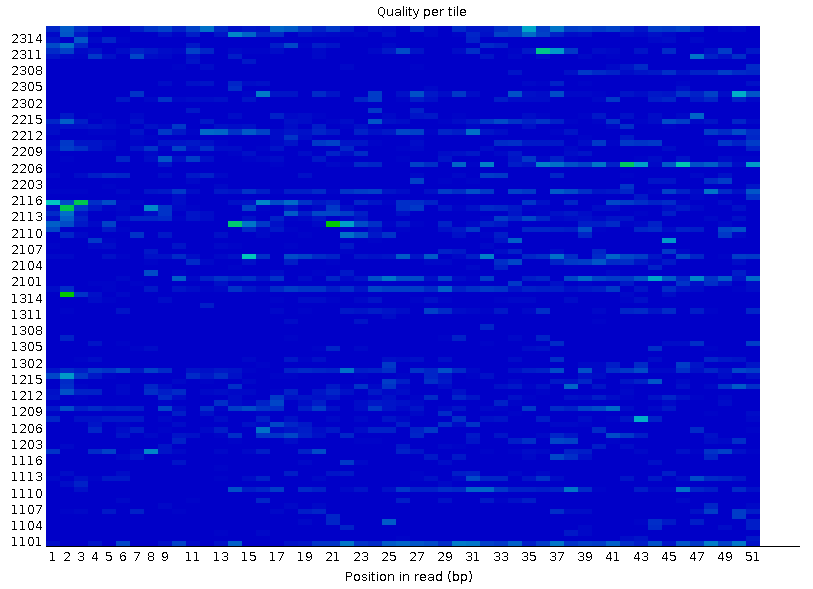 Рисунок 2B. Качество ячеек Иллюмины: вторая реплика |
По оси ординат отложены номера ячеек, по оси абсцисс - номер нуклеотида в риде. Качество сигнала в ячейке тем лучше, чем "холоднее" цвет, идеальной картинкой являлся бы чисто синий фон. Представленный на первой картинке, соответствующий первой реплике красный фрагмент свидетельствует о плохом качестве сигнала - самом плохом. Если сигнал плох, то чаще во многих нуклеотидах одной ячейки - заметны характерные выделяющиеся горизонтальные линии. | |
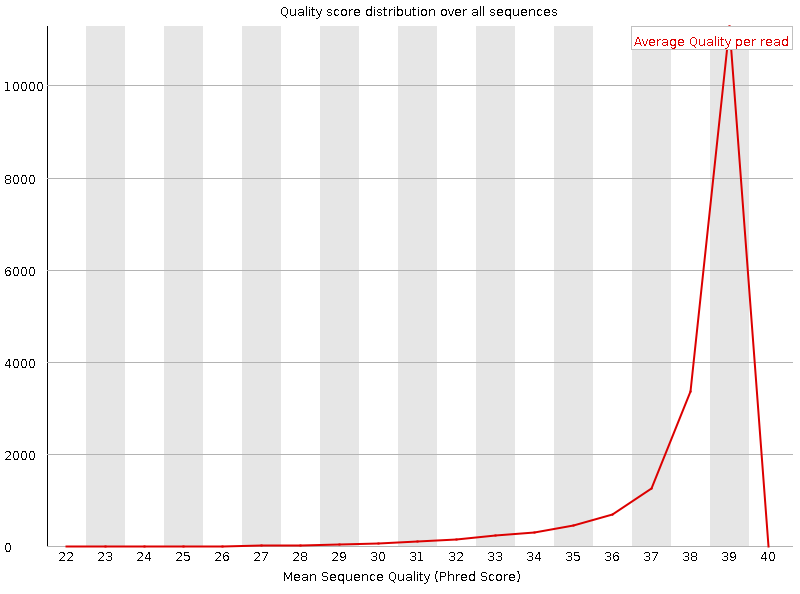 Рисунок 3A. Распределение качества ридов по длине последовательности: первая реплика | 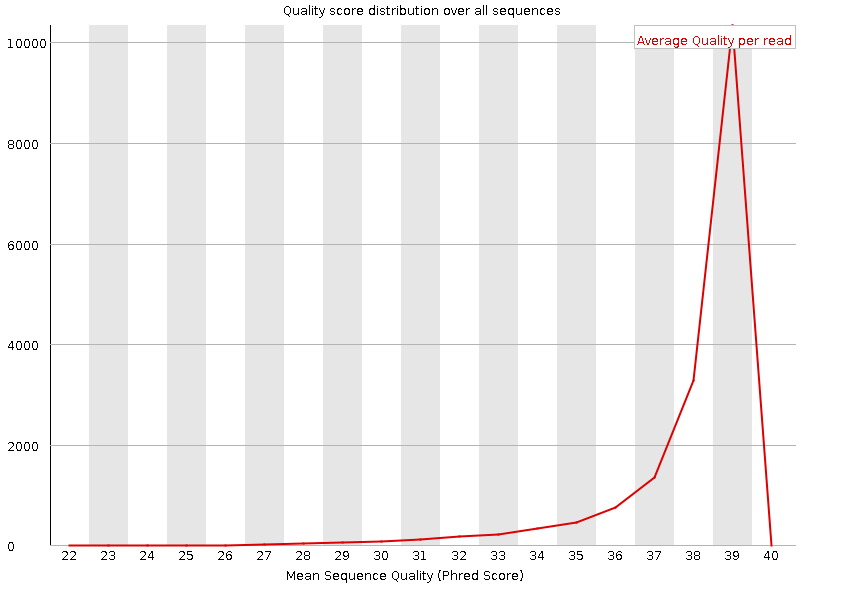 Рисунок 3B. Распределение качества ридов по длине последовательности: вторая реплика |
От стандартной картинки распределения отличается безинтересно: пик шире нормы, стандартное распределение не затрагивает риды так рано, качество повышается в узком диапазоне. | |
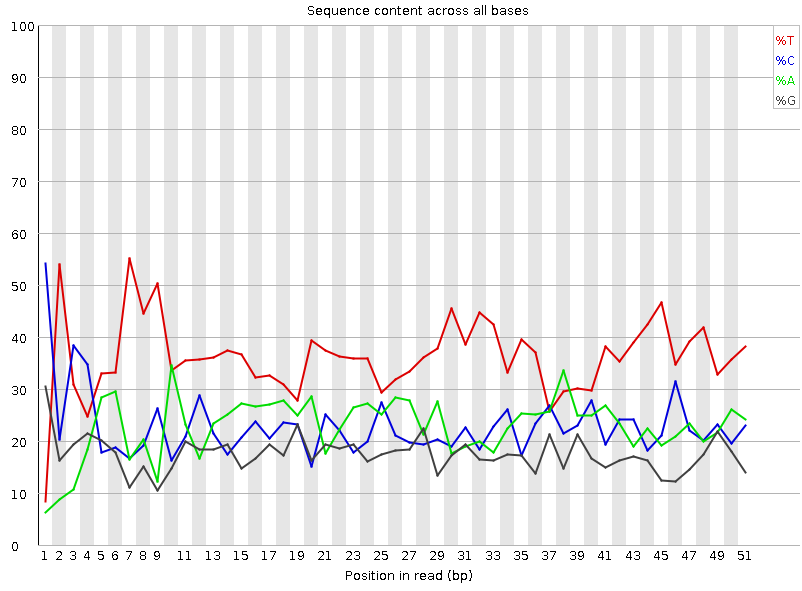 Рисунок 4A. Доля каждого основания на позиции: первая реплика | 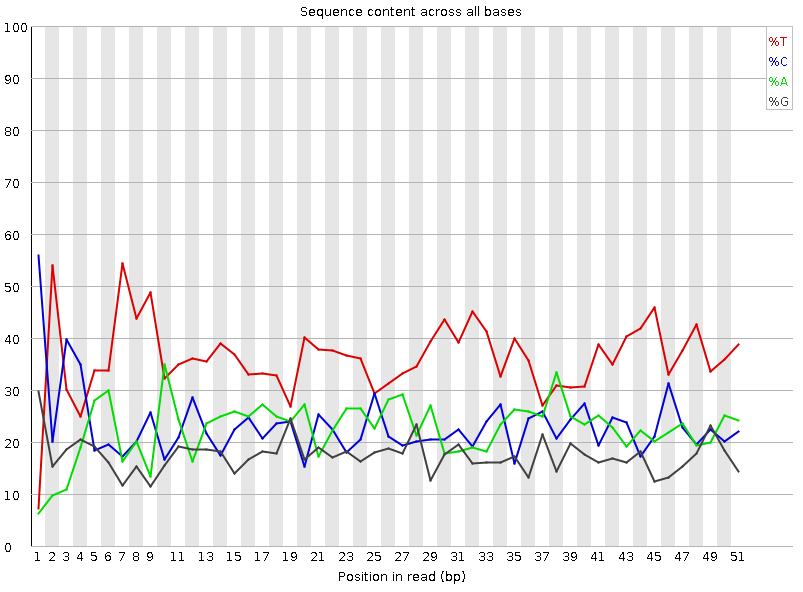 Рисунок 3B. Доля каждого основания на позиции: вторая реплика |
Можем наблюдать рваное пиковатое начало с повышенными значениями для цитозина и тимина, и несколько сдвинутыми отосительно них восходящими пиками их пар. И если для цитозина и гуанина отклонение действительно крошечное, то вот аденин изрядно шалит, поднимая пик очень медленно. Из интересного: в районе 6-9 позиций ридов есть сдвоенный пик тимина, сильно превосхододящий по величине таковые для остальных нуклеотидов примерно в два, а то и в пять раз - для гуанина. Закономерно, ведь на всём протяжении графиков ридов тимина больше всего, что и смущает программу - несоответствие. Причем такие пики наблюдаются на чтениях обоих реплик, что указывает либо на систематическую ошибку методики, либо на какой-то сдвиг в последовательностях. Судить тут уже невозможно. | |
 Рисунок 5A. Распределение GC: первая реплика | 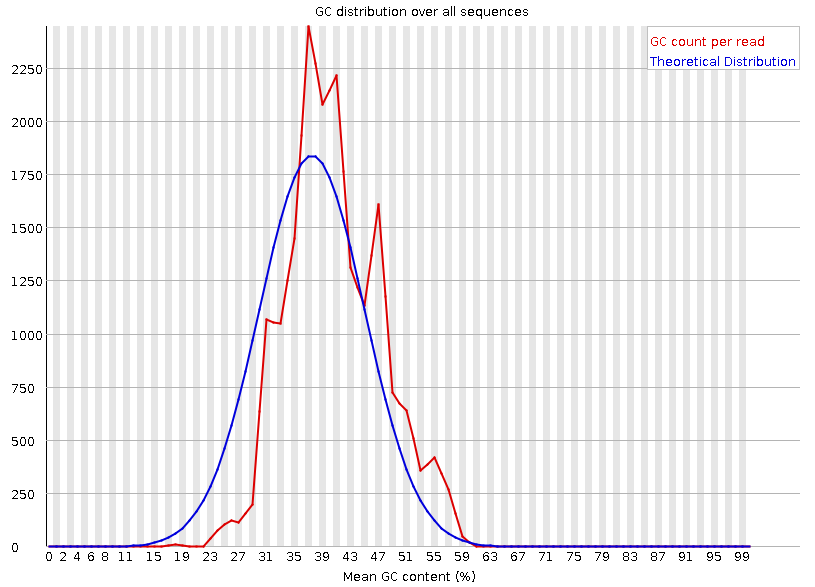 Рисунок 4B. Распределение GC: вторая реплика |
GC=распределение в обоих случаях не совпадает с теоретическим. Помимо этого пик реального распределения несколько сдвинут, так как увеличение доли ПС начинается позже. К тому же наблюдается второй пик на более поздних позициях - с 45 по 49 рид. | |
 Рисунок 6A. Повторение последовательностей: первая реплика | 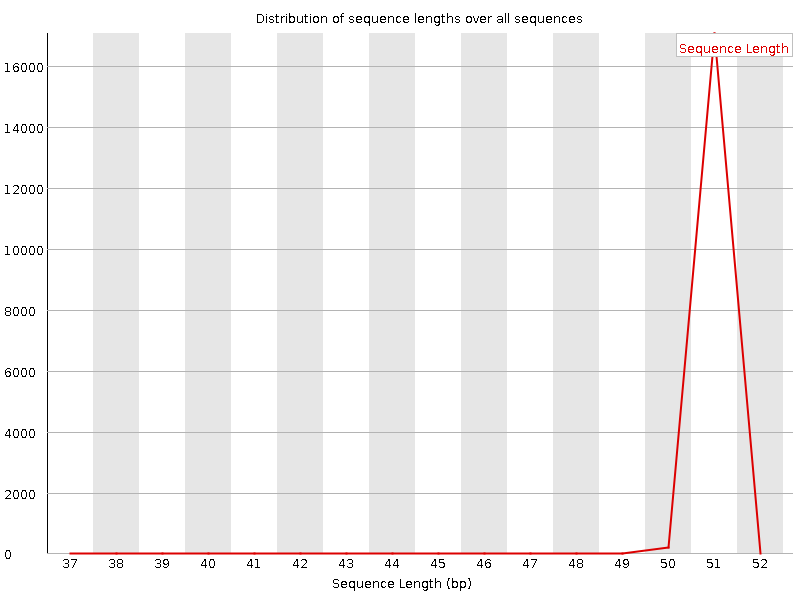 Рисунок 5B. Повторение последовательностей: вторая реплика |
| Очередное распределение по длине, на этот раз -длины ридов по длине последовательностей. Зависит чисто от того, как порезали, и предпочтительно распределение с пиком посередине - совпадающие медиана и средне значение. Здесь это, очевимдно, неверно для обеих реплик: технические особенности. | |
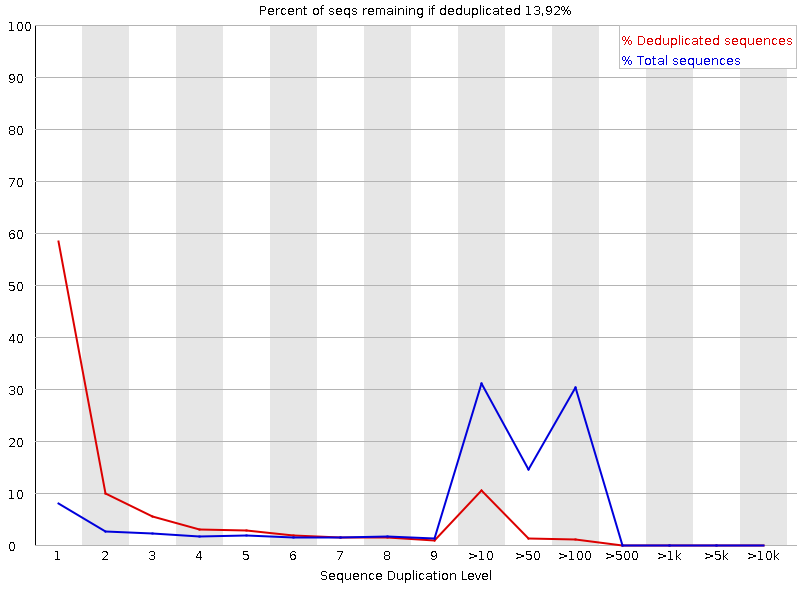 Рисунок 7A. Повторение последовательностей: первая реплика | 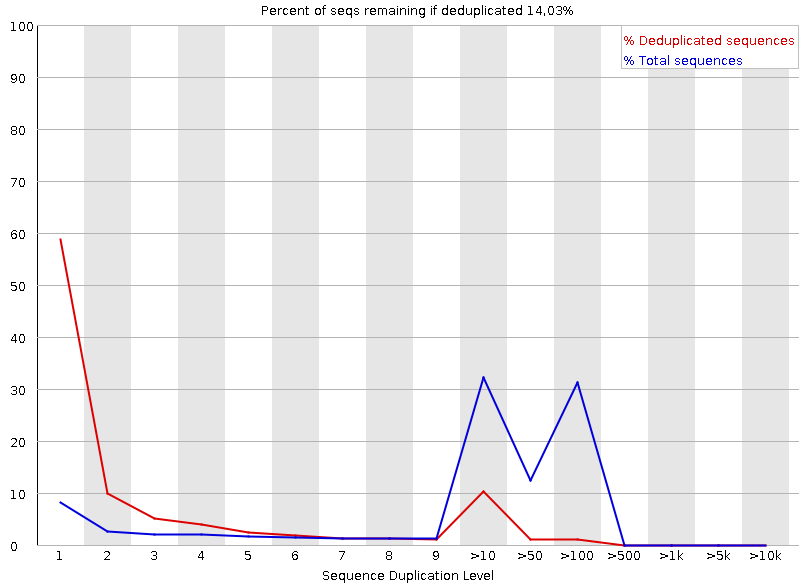 Рисунок 7B. Повторение последовательностей: вторая реплика |
Этот график показывает колчество дуплицированных последовательностей: синяя линия - процент всех последовательностей, красная - дуплицированных образцов, их исходников, без копий. В районе 9->50 - двойной пик, когда как далее - пик лишь для синего. Изначально наблюдалось большое количество дуплицированных - отсюда повшенное количество дуплекантов. Такой большой вклад дуплицируемых последовательностей посередине - крайне нетипичен. | |
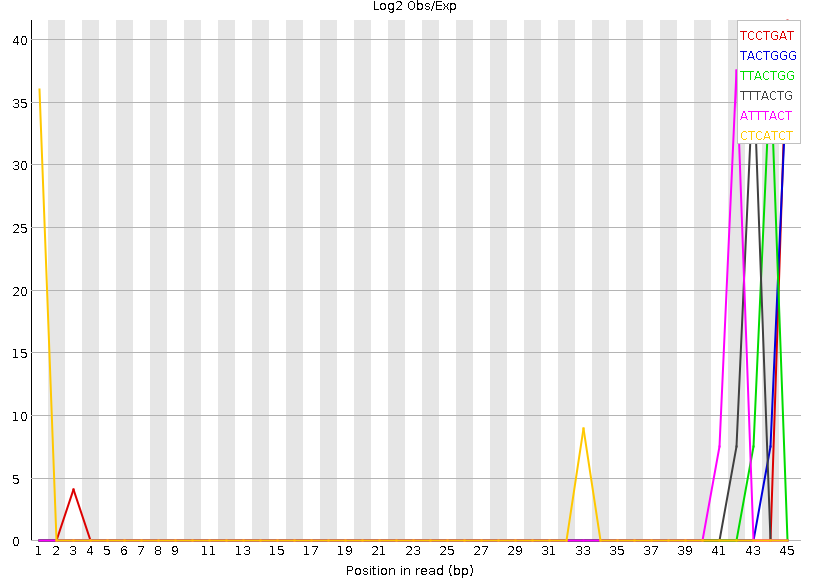 Рисунок 8A. Kmer-контент, график: первая реплика | 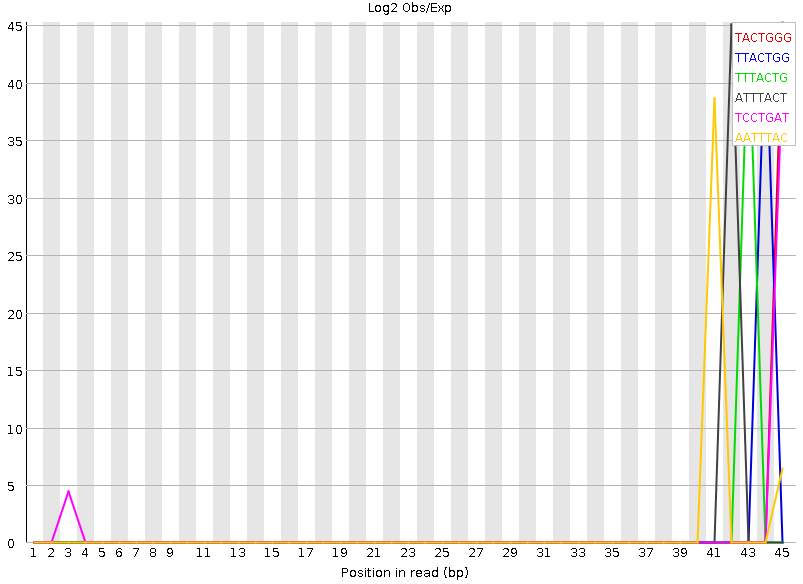 Рисунок 8B. Kmer-контент, график: вторая реплика |
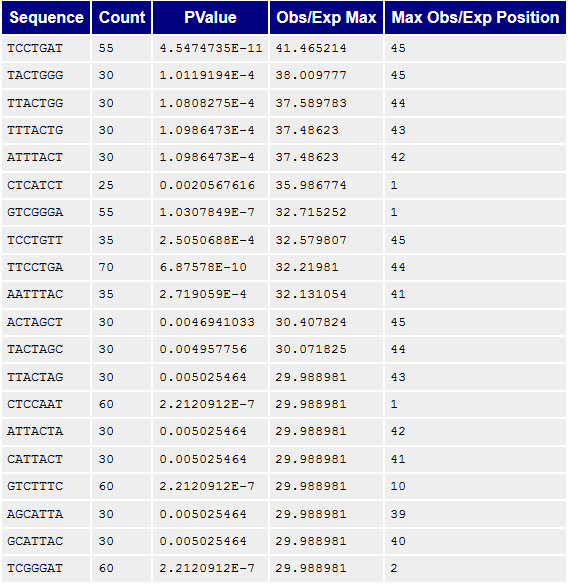 Рисунок 9A. Kmer-контент, последовательности/паттерны: первая реплика | 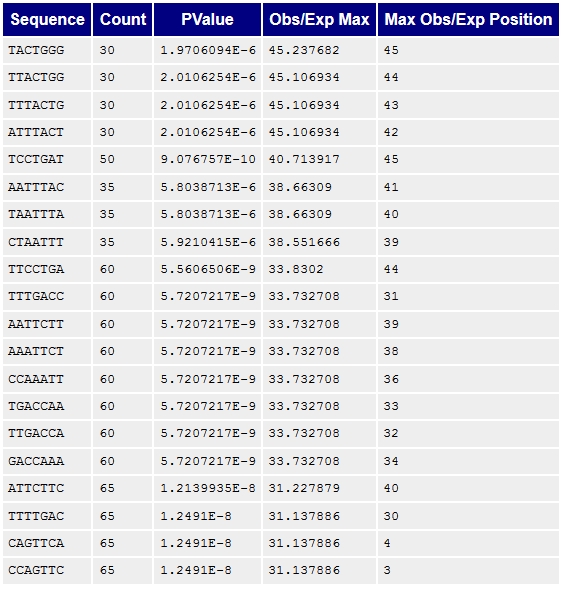 Рисунок 9B. Kmer-контент, последовательности/паттерны: вторая реплика |
Kmer-контент, показывает количество повторяющихся паттернов. Все они достаточно короткие, не больше семи нуклеотидов, поэтому наблюдается большое количество случайных совпадений, которые, по идее, программой воспринимаются как нечто не нормальное. | |
Этап третий: картирование чтений
С помощью программы Hisat2
чтения были откартированы без предварительной очистки, так как очистка бы уничтожила их полностью - последовательности слишком коротки.
Необходимые данные для запуска были перемещены в рабочую директорию и проведён запуск программы:
Индексация референсной последовательности была произведена нами ранее, опять же, в предыдущем практикуме,
а потому мы имеем возможность скопировать восемь файлов вида chr14.*.ht2, где * - цифра от 1 до 8. Построение выравнивания производилось точно так же, как в предыдущем практикуме,
за исключением того, что никак не ограничены возможные разрывы в выравниваниях.
После того, как референсная последовательность была проиндексирована, очищенные чтения были выровнены по индексированной последовательности
- файлам выдачи без подрезания ридов, типичного для концевых участков.
Этап четвёртый: анализ выравниваний
С помощью пакета samtools была произведён анализ выравнивания.
Сначала файл чтений был переведён в формат .bam из формата .sam выдачи hisat (см. предыдущий параграф или предыдущий практикум).
Запуск программы представлен в таблице.
- -b -- выходной файл бинарный
- -o -- указание имени выходного файла
Далее происходила сортировка выравнивание чтений с референсом (получившийся после картирования .bam файл) по координате начала чтения. Опция -Т с последующим указанием файла это операция перенаправения временных файлов.
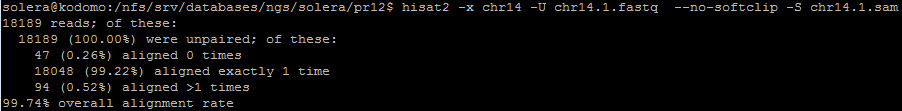
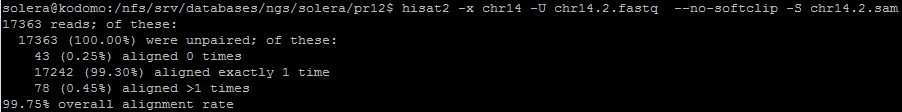
Отсортированный файл .bam далее проиндексирован, с послежующим выяснением, сколько чтений, в итоге, было откартировано на геном: команды можно наблюдать на рисунке ниже.
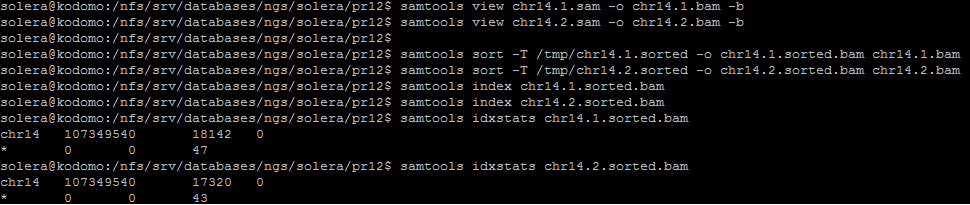
Программа выдала следующий анализ: после названия последовательности идут её длина, число картировавщихся ридов, число некартировавшихся ридов. Можем сравнить данную выдачу с логом программы хисат (Рисунок 10). Действительно, из 18142 картированных по мнению indxstats ридов первой реплики, все упомянуты hisat, а 94, как видно из Рисунка 10, даже прокартировались более одного раза. Что касается второй реплики - всего 17320 картированных по мнению indxstats ридов первой реплики, все упомянуты hisat, а 78 прокартировались более одного раза. Число некартированных так же в обоих случаях совпадает.
Для анализа так же доступен файл chr14.sam - файл выдачи hisat2.
Анализ генов и перекрывающихся с ними прочтений
Этот этап будет так же частично описан в следующем практикуме, ниже приведён более подробный обзор.
По итогам работы программ пакета bedools, которые представлены в таблице ниже, был получин файл, с содержимым которого вы можете ознакомиться по
Б a href='gene.txt'>ссылке. Оказалось, что чтения ложатся на единственный ген HSP90AA1,
кодирующий беллк теплового шока.
По данным NCBI от 21 ноября 2017, данные HSP90AA1 были собраны в таблицу:
| Размер | Координата начала | Координата конца | Количество экзонов | Покрытие | Направление | Локация | Референсы по данным HPA | Дополнительная информация |
| 59,022 bases | 102,080,738 bp from pter | 102,139,759 bp from pter | 13 | 2624 | minus | 14q32.31 | Cancer-related genes Plasma proteins Predicted intracellular proteins | Белок, кодируемый этим геном, это молекулярный шаперон, который функционирует как гомодимер. Закодированный белок способствует правильному складыванию конкретных белков-мишеней с использованием активности АТФазы, которая модулируется ко-шаперонами. Для этого гена были найдены два варианта транскрипции, кодирующих различные изоформы. [предоставлено RefSeq, январь 2012 г.] |
Отдельно хотелось бы сказать про подсчёт покрытий - подсчет осложнился тем, что многие строки повторялись и перекрывались. Поэтому пришлось сконструировать новый файл, выданному с опциями '-wa и -wb', в этом случае считалось количество строк, содержащих определенный ген. Каждая строка соответствует покрытию ридом одного участка Однако в самой разметке имеются повторы. Так что в таблице приведены оценки сверху, потому как в реальности при обсчёте покрытий для генов может оказаться чуть меньше, раз уж даже так файл выдаёт повторы. Подсчёт проводился по файлу без нулей, с результатами в файле Excel вы можете ознакомиться, скачав файл.
