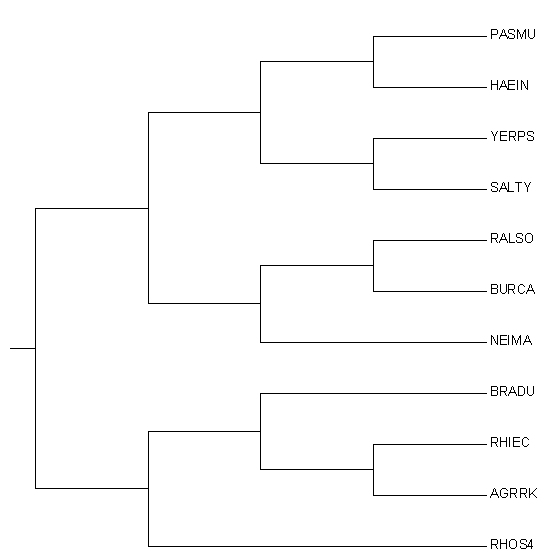
Построение дерева
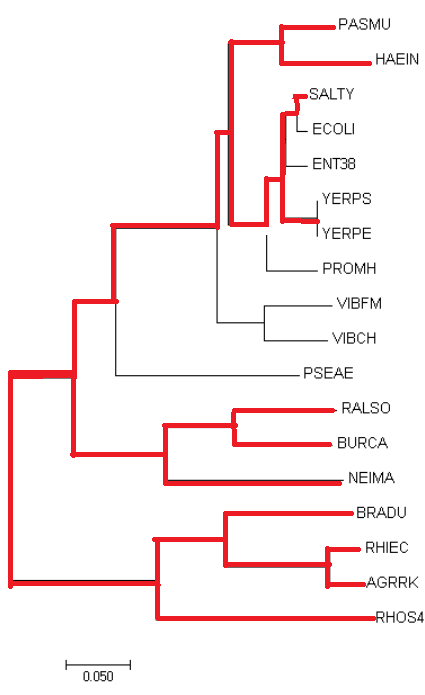 Рисунок 2.Исходное дерево
Рисунок 2.Исходное дерево
|
Дерево строилось по "матрице" исходного дерева. На Рис.2 красным выделены все ветви, которые были сохранены в конечном филогенетическом отношении.
Каждая развилка переходит на новый рисунок развилкой, ломаная линия трансформируется в прямую.
На рисунке мы можем наблюдать, что две нижние группы: {BRADU,RHIEC,AGRRK,RHOS4} и {RALSO,BURCA,NEIMA} полностью переходят в новое дерево,
когда как оставшаяся обширная группа переходит не полностью. В "верхней" кладе сохраняется только три узла, притом, что сохраняется исходный
{PASMU,HAEIN,YERPS,SALTY} против {RALSO,BURCA,NEIMA}, далее по ветви {PASMU,HAEIN,YERPS,SALTY} находятся узлы {PASMU,HAEIN} против {YERPS,SALTY}.
Ветвь {PASMU,HAEIN} имелась на исходном дереве, и там же противолежала ветви, из которой сохраняются лишь бактерии YERPS и SALTY.
Раннее ветвь, которой они были объеденены, имела две нетривиальные ветви в составе (что плохо видно на рисунке) - собственно, {SALTY, ECOLI, ENT38}
против {YERPS, YERPE}, теперь же это отношение между YERPS и SALTY сохранилось в виде ветви с двумя соответствующими листами.
Укоренение программа MEGA, с помощью которой была реализована визуализация деревьев, построила самостоятельно, как и ранее, между двумя обширными ветвями,
одна из которых в новое дерево перешла полностью ({BRADU,RHIEC,AGRRK,RHOS4}), а вторая - частично ({PASMU,HAEIN,YERPS,SALTY,RALSO,BURCA,NEIMA}).
Таким образом, очевидно, что построение нового дерева верное.
|
Таксономическое положение бактерий
Было установлено в соответствии с данными таксономического браузера NCBI.
Ниже приведена сводная таблица с данными, касательно каждой из бактерий.
Нетривиальные ветви, которые мы могли видеть на сконструированном заранее дереве, однозначно выделяют нам следующие таксономические группы:
| Бактерия |
Организация |
Царство |
Тип |
Класс |
Отряд |
Семейство |
Группа |
Род |
Комплекс |
| Pasteurella multocida |
cellular organisms |
Bacteria |
Proteobacteria |
Gammaproteobacteria |
Pasteurellales |
Pasteurellaceae |
- |
Pasteurella |
- |
| Haemophilus influenzae |
cellular organisms |
Bacteria |
Proteobacteria |
Gammaproteobacteria |
Pasteurellales |
Pasteurellaceae |
- |
Haemophilus |
- |
| Yersinia pseudotuberculosis |
cellular organisms |
Bacteria |
Proteobacteria |
Gammaproteobacteria |
Enterobacterales |
Yersiniaceae |
- |
Yersinia |
Yersinia pseudotuberculosis complex |
| Salmonella typhimurium = Salmonella enterica subsp. enterica serovar Typhimurium |
cellular organisms |
Bacteria |
Proteobacteria |
Gammaproteobacteria |
Enterobacterales |
Enterobacteriaceae |
- |
Salmonella |
Salmonella enterica(subsp. enterica) |
| Ralstonia solanacearum |
cellular organisms |
Bacteria |
Proteobacteria |
Betaproteobacteria |
Burkholderiales |
Burkholderiaceae |
- |
Ralstonia |
- |
| Burkholderia cenocepacia |
cellular organisms |
Bacteria |
Proteobacteria |
Betaproteobacteria |
Burkholderiales |
Burkholderiaceae |
- |
Burkholderia |
Burkholderia cepacia complex |
| Neisseria meningitidis |
cellular organisms |
Bacteria |
Proteobacteria |
Betaproteobacteria |
Neisseriales |
Neisseriaceae |
- |
Neisseria |
- |
| Bradyrhizobium diazoefficiens |
cellular organisms |
Bacteria |
Proteobacteria |
Alphaproteobacteria |
Rhizobiales |
Bradyrhizobiaceae |
- |
Bradyrhizobium |
- |
| Rhizobium etli |
cellular organisms |
Bacteria |
Proteobacteria |
Alphaproteobacteria |
Rhizobiales |
Rhizobiaceae |
Rhizobium/Agrobacterium group |
Rhizobium |
- |
| Agrobacterium tumefaciens |
cellular organisms |
Bacteria |
Proteobacteria |
Alphaproteobacteria |
Rhizobiales |
Rhizobiaceae |
Rhizobium/Agrobacterium group |
Agrobacterium |
Agrobacterium tumefaciens complex |
| Rhodobacter sphaeroides |
cellular organisms |
Bacteria |
Proteobacteria |
Alphaproteobacteria |
Rhodobacterales |
Rhodobacteraceae |
- |
Rhodobacter |
- |
Таблица 2. Полное таксономическое положение
|
Наглядная реконструкция филогении
Теперь найдём общие таксоны у всех вышеперечисленных бактерий.
Очевидно, все они относятся к типу Proteobacteria. Далее мы можем наблюдать членение по нетривиальным ветвям:
- семейство Pasteurellaceae - ветвь {PASMU,HAEIN}
- отряд Enterobacterales - ветвь {YERPS,SALTY}
- класс Gammaproteobacteria - ветвь {PASMU,HAEIN,YERPS,SALTY}
- семейство Burkholderiaceae - ветвь {RALSO,BURCA}
- класс Betaproteobacteria - ветвь {RALSO,BURCA,NEIMA}
- группа Rhizobium - ветвь {RHIEC,AGRRK}
- отряд Rhizobiales - {BRADU,RHIEC,AGRRK}
- класс Alphaproteobacteria - {BRADU,RHIEC,AGRRK,RHOS4}
Так же очевидно, что группы альфа- и бетапротеобактерии более сближены относительно гаммапротебактерий (нетривиальная ветвь {PASMU,HAEIN,YERPS,SALTY,RALSO,BURCA,NEIMA} против {BRADU,RHIEC,AGRRK,RHOS4}).
Реконструкция филогении по белкам семейства факторов высвобождения пептидной цепи RF1
Из базы данных Uniprot были получены последовательности белков семейства RF1 для вышеперечисленных видов бактерий: bact.fasta.
С помощью сервиса Muscle на сайте EMBL-EBI было построено выравнивание: align.fasta.
Уже на странице сервиса Muscle можно было наблюдать безрадостную картину несовпадения дерева автоматической выдачи с нашим деревом:
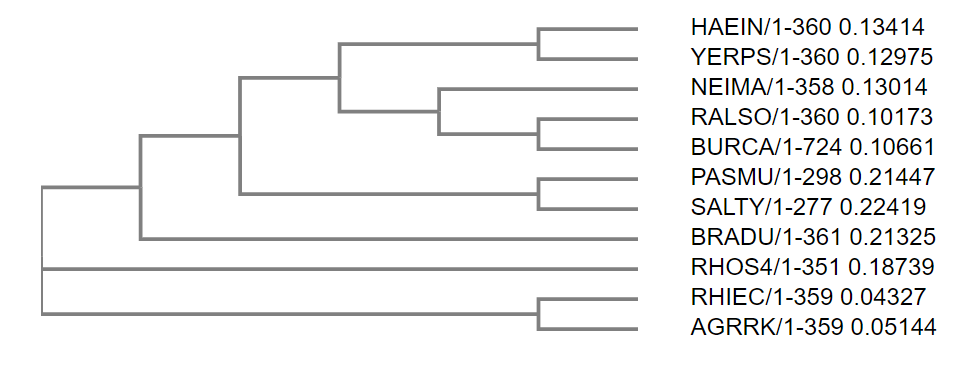 (((((RALSO:0.10521,BURCA:0.10313):0.10289,(HAEIN:0.13570,YERPS:0.12819):0.08045):0.02658,(PASMU:0.22336,SALTY:0.22682)
:0.38489):0.02344,BRADU:0.21151):0.02049,RHOS4:0.18442,(RHIEC:0.04284,AGRRK:0.05187):0.11866);
(((((RALSO:0.10521,BURCA:0.10313):0.10289,(HAEIN:0.13570,YERPS:0.12819):0.08045):0.02658,(PASMU:0.22336,SALTY:0.22682)
:0.38489):0.02344,BRADU:0.21151):0.02049,RHOS4:0.18442,(RHIEC:0.04284,AGRRK:0.05187):0.11866);
В программе MEGA по полученному выравниванию тремя различными способами была реконструирована филогения:
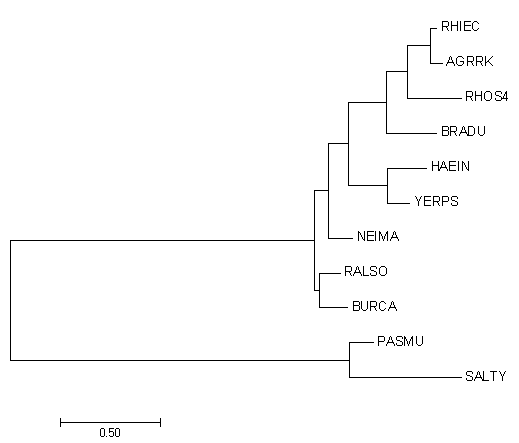 Рисунок 3. Maximum Likelyhood tree Рисунок 3. Maximum Likelyhood tree |
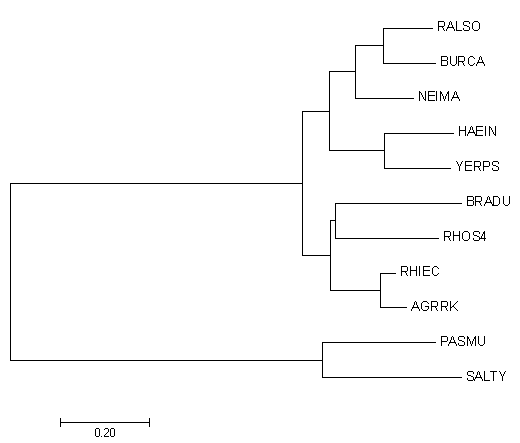 Рисунок 4. Neighbor-Joining tree Рисунок 4. Neighbor-Joining tree |
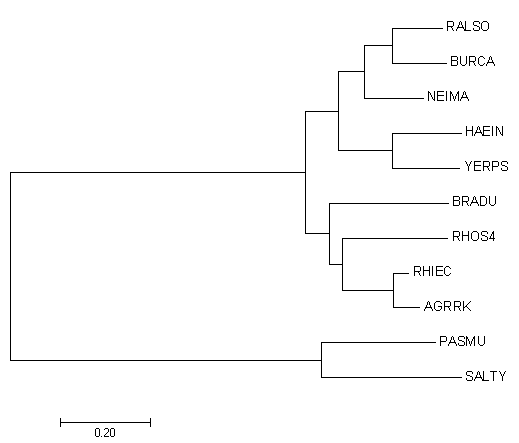 Рисунок 5. Minimum-Evolution tree Рисунок 5. Minimum-Evolution tree |
Соответственно, новые деревья существенно отличаются от дерева, построенного нами ранее. Между собой они так же не идентичны,
хотя есть и схожие черты, особенно между двумя последними.
С выравниванием, по которому строились эти деревья, можно ознакомиться по ссылке.
| Интересная, сопутствуящая выдаче рисунков информация от MEGA |
| The evolutionary history was inferred by using the Maximum Likelihood method based on the JTT matrix-based model. The tree with the highest log likelihood (-4131.64) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using a JTT model, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 10 amino acid sequences. All positions containing gaps and missing data were eliminated. There were a total of 264 positions in the final dataset. Evolutionary analyses were conducted in MEGA7.
|
The evolutionary history was inferred using the Neighbor-Joining method. The optimal tree with the sum of branch length = 3.39202580 is shown. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Poisson correction method and are in the units of the number of amino acid substitutions per site. The analysis involved 10 amino acid sequences. All positions containing gaps and missing data were eliminated. There were a total of 264 positions in the final dataset. Evolutionary analyses were conducted in MEGA7.
|
The evolutionary history was inferred using the Minimum Evolution method. The optimal tree with the sum of branch length = 3.38593591 is shown. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Poisson correction method and are in the units of the number of amino acid substitutions per site. The ME tree was searched using the Close-Neighbor-Interchange (CNI) algorithm at a search level of 1. The Neighbor-joining algorithm was used to generate the initial tree. The analysis involved 10 amino acid sequences. All positions containing gaps and missing data were eliminated. There were a total of 264 positions in the final dataset. Evolutionary analyses were conducted in MEGA7.
|
Дополнительное задание. Работа через fprotdist и fneighbor.
Вернуться назад
На главную страницу
©Solonovich Vera,2017