Алгоритм PSI-BLAST (Position-Specific Iterated BLAST) предназначен для поиска удаленных гомологов белков. Для реализации этого алгоритма используется техника создания PSSM(Position-Specific Scoring Matrix) - матриц, в которых вес замены аминокислотного остатка зависит от его положения в выравнивании. Аминокислоты в строках соответствуют номерам позиций тех в выравнивании в столбцах. Значение в ячейке таблицы характеризует вес данной аминокислоты в данной позиции, и чем чаще та встречается в этом месте в гомологах последовательности, тем больше ее вес. Таким образом, в ячейках PSSM-матрицы стоят положительные или отрицательные целые числа. Положительные указывают, что аминокислота замещается чаще, чем можно ожидать в случайной модели. Отрицательные значения - наоборот.
PSSM можно использовать для поиска новых гомологов, реализуя итеративность алгоритма, которые подразумевает корректировку профиля после выполнения каждого нового шага в соответствии с новыми данными. Это реализует поиск, аналогичный поиску через матрицу BLOSUM в классическом алгоритме BLAST, где PSSM позволяет оценить вес выравниваний новых последовательностей с исследуемой, выявив лучшие находки. После обычного белок-белкового бласта программа на основе выдачи строит профиль PSSM на основе множественного выравнивания находок с E-value < 0.005 по дефолту, после чего новые последовательности с E-value < 0.005 добавляются в выравнивание, по которому строится новая PSSM, и так далее, пока поиск не "сойдётся" - список находок после последней итерации не изменится.
Для тестирования вышеописанного алгоритма был выбран белок O05886 - фактор гибернации рибосомы, кодируемый геном hpf в Mycobacterium tuberculosis (strain ATCC 25618 / H37Rv), известной более в рускоязычной литературе как палочка Коха, возбудитель туберкулёза.
Данный белок является необходимым фактором димеризации рибосомных субъединиц при иницации трансляции бактерии, активизируя 70S рибосомы собираться в комплекс 100S с 30S на первых этапах процесса синтеза белка.
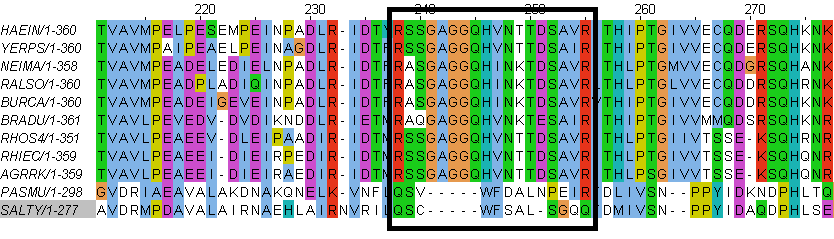
Был проведён поиск PSI-BLAST по базе данных SwissProt, с пороговым значением E-value = 0.001. Ниже представлена таблица итераций поиска:
| Таблица 1. Итерации поиска | |||||
|---|---|---|---|---|---|
| Номер итерации | Число находок выше порога 0,001 | Идентификатор худшей находки выше порога | E-value этой находки | Идентификатор лучшей находки ниже порога | E-value этой находки |
| №1 | 31 | P28613.2 | 6e-04 | P26983.1 | 0.001 |
| №2 | 46 | P71346.3 | 5e-11 | P9WMA8.1 | 0.009 |
| №3 | 51 | P24694.1 | 2e-23 | P9WMA8.1 | 0.002 |
| №4 | 51 | P24694.1 | 5e-24 | P9WMA8.1 | 0.002 |
| №5 | 53 | P24694.1 | 4e-24 | P9WMA8.1 | 0.001 |
| №6 | 53 | P24694.1 | 5e-24 | P9WMA8.1 | 0.001 |
| №7 | 53 | P24694.1 | 4e-24 | P9WMA8.1 | 0.001 | и далее те же самые находки |
Низкое значение надпорогового e-value говорит об относительной консервативности данной группы белков - гомологов среди различных организмов. Разница между E-value хороших и плохих находок составляет 21 порядков, что свидетельствует об очень хорошей. Большинство находок совпадает по функции с исходным белком. Отличия составляют только P9WMA8.1 - другой белок палочки Коха, вовлечённый в регуляцию трансляции, другой P71346 - YFIA_HAEIN, фактор ассоциациированный с рибосомой Y возбудителя пневмонии, который предотвращает димеризацию рибосомной субъединиц 70S, возможно, из-за этого имея гомологичны участок последовательности с исходным нашим белком.
[ARH]-[STA]-x-G-x-G-G-Q-[HNGCSY]-[VI]-N-x(3)-[ST]-[AKG]-[IV] |
R-[SA]-x-G-A-G-G-Q-H-[VI]-N-x-T-[DE]-S-A-[IV]-R-[ILV]-T-H-[IL]-P-[TS]-G |
| В базе данных далее были найдены все соответствия паттерну из банка Swiss-Prot: было найдено 419 совпадений по данному паттерну, когда как на сайте Uniprot через поиск по мнемоникее rf1_* - 408, то есть на 10 больше. С помощью средств Python было рассчитано количество истинных находок (True positives, TP), число ложных находок (False positives, FP), и число ненайденных находок (False negatives, FN) - TP нашли, как пересечение списков, FP - как оставшиеся в списке для модифицированного паттерна идентификаторы, FN - как оставшиеся в листе uniprot. |
| ||||||||||||||||||
|
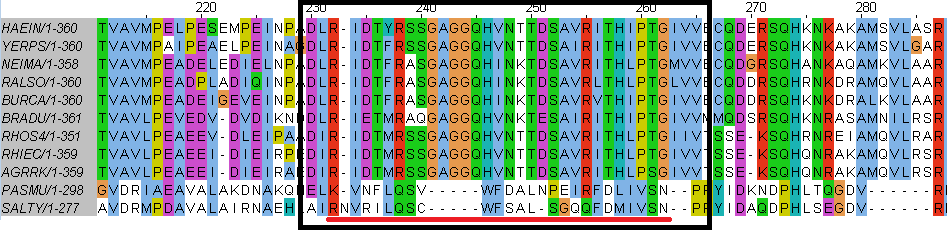
Как мы можем наблюдать по результатам выдачи, ненайденных белков не слишком много, те составляют меньше четверти находок, что позволяет нам считать отредактированный паттерн хорошим. Попытаемся изменить паттерн, чтобы достоверность модификации его повысилась. Сначала попробуем ограничить паттерн его прошлыми границами, убрав дополнительные колонки. |
| ||||||||||||||||||
|
Соотношение верных находок к неверным немедленно возросло. Причём, судя по следующим тестам, все из которых я приводить ниже не буду, увеличение исходного паттерна вообще не помогает повысить число положительных находок, отсеивает большее количество не найденных, конечно, но пропорция между ними не сохраняется исходной и свидетельствует об уменьшении количества обнаруженных относительно не обнаруженных: |
| ||||||||||||||||||
|
Теперь попытаемся в новых рамках паттерна что-нибудь изменить, чтобы понять, помогает ли нам его уточнение относительно первоначальных рамок. Новое задание букв более обширное, вместо общности [DE] далее была выбрана любая буква, например, а если. Продолжить в том же духе - интересно, что отдельна замена T в тринадцатой позиции не привела к улучшению результата, а замена A в пятой позиции на общую x уменьшила FN и увеличила TP значения, но и замена обоих частностей сделала то же самое. Ниже представлен финальный и самый лучший результат. |
| ||||||||||||||||||
| Таблица 2E. Итоги анализа находок | |
|---|---|
| R-[SA]-x-G-x-G-G-Q-H-[VI]-N-x-T-x-S-A-[IV] R-[SA]-x-G-x-G-G-Q-H-[VI]-N-x(3)-S-A-[IV] | |
| FN | 57 |
| FP | 224 |
| TP | 351 |
| TP/FN | 6.15 |
| TP/SUM | 0.555 |
| FP/SUM | 0.355 |
| FN/SUM | 0.09 |
Так как задача стояла выбрать наилучший паттерн для нашего случая, выбираем наиболее конкретный. Итого: - наилучший модифицированный паттерн.
На главную страницуВернуться назад
©Solonovich Vera,2017