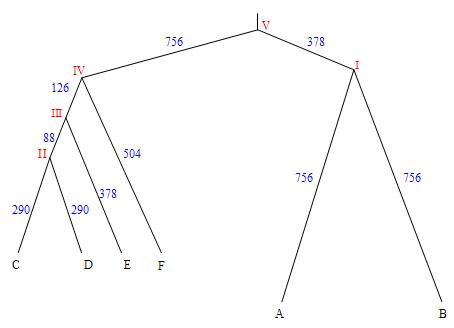
Оно является укоренённым, т.к. в корне (обозначено V) находится последовательность гена белка rho_ecoli, в которую были внесены мутации с учётом приведённых здесь эволюционных расстояний (они отмечены синим), рассчитанных на длину гена. В результате были получены последовательности, отвечающие листьям дерева, как то: А, B, C, D, E, F.
Далее для полученных результатов было построено множественное выравнивание, включающее в себя 6 последовательностей. Задача теперь заключалась в обратной процедуре: восстановить дерево и попробовать найти предковую последовательность.
Для проведения бутстреп–анализа в пакете Phylip (the PHYLogeny Inference Package) можно последовательно использовать 3 программы: seqboot, dnaml, consense. Исходящий файл одной программы является входящим для другой, а поэтому файлы нужно переименовывать в соответствии с особенностями каждой программы. Сначала выравнивание было переведено в Phylip–формат: с помощью программы FAR каждая последовательность располагается на одной строчке так, что первые 10 символов отводятся под название. Если название слишком короткое, то в оставшемся пространстве оставляют пропуски. Сами последовательности должны иметь одинаковую длину. Это важно для того, чтобы в итоге получить корректные результаты. Над выравниванием проставляются 2 значения: число последовательностей и длина последовательностей. Но выравнивание в этом формате можно получить и с помощью других программ, например, ClustalW или TreeView. Важно, чтобы получившийся текстовый файл не содержал непечатуемых знаков.
Программы пакета Phylip при запуске начинают искать входящие файлы с определённым именем, например, infile. Если такого не обнаружилось, программа попросит ввести имя вручную.
Метод Bootstrap заключается в проведении анализа множества последовательностей, как бы развивающихся независимым образом. Множество бутстреп–реплик выравнивания можно получить с помощью программы seqboot пакета Phylip. В алгоритме программы заложены 4 метода: Bootstrap, Jackknife, Permute, Rewrite, которые выбираются командой J в командной строке. Количество реплик задаётся с помощью команды R, после чего вводится натуральное число. Наиболее адекватным интервалом является 100 — 1000 реплик, в зависимости от поставленной задачи. В нашем случае это — 100. В исходящем файле outfile получаются 100 выравниваний, по 6 последовательностей в каждом, причём с мутациями: где–то пропущен нуклеотид, где–то удвоен, но при этом длина остаётся постоянной — она не изменяется: если во входящем файле длина у нас 1260 н.о., то такая же и после работы программы.
Получившийся в результате файл outfile был переименован в infile с целью подать его на вход другой программе dnaml, которая использует скрытую Марковскую модель для символьно–ориентированной реконструкции филогенетического дерева. Каждая линия здесь эволюционирует независимо. Интерфейс у всех этих программ аналогичный, и количество выравниваний, которое нужно обработать, задаётся командой M, при этом программа спросит, какой тип входящих данных использовать (множественный набор данных, наверное, имеется в виду множественное выравнивание; или множественные веса), потом нужно задать собственно объём данных (т.е. 100) и число, называемое random number seed, — оно используется при статистических расчётах, может принимать какое–либо значение из интервала от 1 до 32767, но должно отвечать формуле 4n+1, т.е. быть нечётным. Если задать чётное, программа попросит ввести random number seed заново. Меняя это число и перезапуская программу, можно получать и более хорошие деревья. Ещё необходимо ввести количество перестановок, которое будет произведено с последовательностями в пределах каждого выравнивания в наборе из 100 выравниваний. Порядок, в котором идут последовательности одна за другой, играет роль при поиске различных деревьев. Описывая топологию корневого дерева с 6 "листами", мы будем иметь 4 различных разбиения множества листьев. Но так как программа нам здесь построит неукоренённое дерево, то остаётся три различных разбиения, что и было введено как число перестановок. В итоге получилось консенсусное (небинарное) дерево вида:
+--------------------------->F.fasta
|
| +------>B.fasta
| +-------100.0-|
| | +------>A.fasta
+------|
| +------------->E.fasta
+-94.0-|
| +------>C.fasta
+-81.0-|
+------>D.fasta
Ради интереса, было построено дерево с использованием таких же параметров программ, но с числом перестановок 4:
+------------->E.fasta
+-94.0-|
| | +------>C.fasta
| +-81.0-|
+------| +------>D.fasta
| |
| | +------>A.fasta
| +-------100.0-|
| +------>B.fasta
|
+--------------------------->F.fasta
Результат, как видно, оказался тем же, только порядок следования немного изменился. Числа на ветвях обозначают сколько было произведено всевозможных разбиений листьев в 100 различных деревьях относительно данной ветви. Если считать эти значения внутренних ветвей за эволюционные расстояния, то они даже в пропорции отличаются от значений на исходном дереве. Но по сравнению с исходной моделью, получившееся дерево оказалось таким же по топологии, единственное отличие — оно неукоренённое!
Как уже указывалось выше, в алгоритме программы seqboot заложены 4 метода:
- Bootstrap — основан на том, что все линии эволюционируют независимо друг от друга; но этот метод подходит далеко не для всех случаев (есть разновидности);
- Jackknife — случайным образом выбирается половина символов в последовательности, остальная же половина выбрасывается. Таким образом данные получаются урезанными вдвое (есть разновидности);
- Permute — дословно означает перестановку: это могут быть столбцы матрицы данных или же отдельные буквы последовательности, в зависимости от разновидности;
- Rewrite — простое переписывание исходных данных в различные форматы, например, XML, NEXUS, PHYLIP.
+--------------------------->F.fasta
|
| +------>B.fasta
| +-------100.0-|
| | +------>A.fasta
+------|
| +------>D.fasta
| +-82.0-|
+-89.0-| +------>C.fasta
|
+------------->E.fasta
remember: this is an unrooted tree!
Это дерево по топологии похоже на предыдущее, а значения внутренних ветвей и порядок листьев отличается. Небольшой вывод: урезание данных вдвое для последовательностей длиной по крайней мере порядка 3 практически никак не влияет на реконструкцию филогении.
Ранее методом Neighbor-Joining было получено дерево:
+-----C.fasta
+--3
! +------D.fasta
!
--4------E.fasta
!
! +-------------A.fasta
! +------------------1
+--2 +-------------B.fasta
!
+---------F.fasta
remember: this is an unrooted tree!
Скобочная формула:
Скобочная формула его была использована для визуализации дерева с помощью программ пакета Phylip в неукорнённом виде и в виде филограммы, укоренённой в среднюю точку.
Неукоренённое дерево можно получить, если подать скобочную формулу на вход программе drawtree, т.е. переписать её в файл intree. Вот результат, переведённый в jpg-формат:
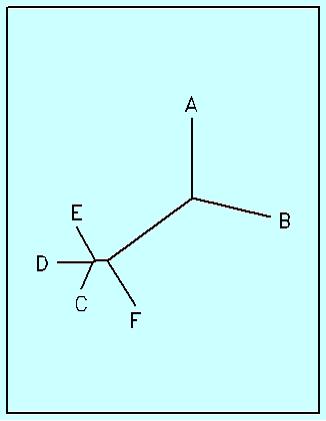
Здесь есть точка, из которой, может показаться, выходит три ветви: это не так, просто ветвь, соединяющая кластер (C;D) с E очень короткая, в скобочной формуле её длина всего 2 сотых, а программа, как видно, соблюдает масштаб. Но в принципе, это более–менее отвечает нашей эволюционной модели.
Потом, чтобы переукоренить дерево в среднюю точку, сначала использовалась программа retree, которой также подавалась скобочная формула. Потом — программа drawgram, у которой по умолчанию параметры стоят так, чтобы была нарисована прямоугольная филограмма, ориентированная горизонтально. Резальтат:
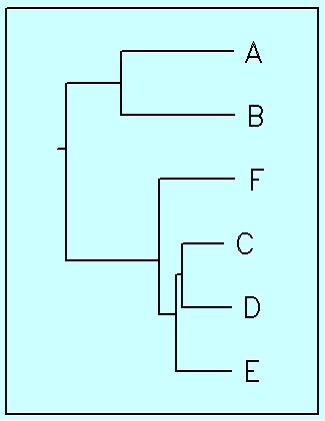
Понятно, что если дерево было предварительно укоренено в среднюю точку, то полученная филограмма с первого взгляда скорее похожа на кладограмму, однако ветви соответствуют здесь эволюционным расстояниям, и это дерево — ультраметрическое.
Для поиска предковой последовательности может быть использована также программа dnaml, которая указывалась выше. Здесь командой 5 можно установить опцию реконструкции гипотетической последовательности. В итоге получилось такое дерево:
+----------------------->B.fasta | | +--------->F.fasta | | 1---------------------4 +------>E.fasta | | +--3 | +---2 +------>D.fasta | | | +----->C.fasta | +--------------------->A.fasta remember: this is an unrooted tree!
В исходящем файле есть также таблица всех последовательностей, которые отвечают не только листьям, но и узлам дерева. Корень по идее должен находиться где–то между 1 и 4 узлами, но он не отмечен. Если принять за корень 1, то он явно не годится под роль "предка", потому что соответствующая последовательность, во–первых, имеет сходство менее 50% с листьями дерева, а во–вторых, совсем не похожа на исходный ген белка rho_ecoli. Выравнивание гипотетической предковой последовательности и реального гена с помощью программы needle.