Алгоритмы и программы множественного выравнивания
Сравнение результатов выравнивания одних и тех же последовательностей тремя разными программами
Для выполнения данного пункта были выбраны последовательности того же семейства белковых доменов, которое уже было рассмотрено в предыдущем практикуме (ABC-транспортёрам). Для их выравнивания использовались программы Muscle, Mafft и T-Coffee (проект в Jalview со сравниваемыми выравниваниями). В качестве референсной программы для выравнивания выступал Muscle, с ним сравнивались все остальные. Для сравнения выравниваний был использован код, написанный моей однокурсницей Еленой Гончаровой (на момент написания данного текста уже известно, что эта программа не находит единичные совпадающие колонки, поэтому далее этот пункт при анализе опускается).
| Muscle | Mafft | Длина |
|---|---|---|
| Совпадающие участки | ||
| 13-31 | 20-38 | 19 |
| 198-199 | 315-316 | 2 |
| 230-261 | 348-379 | 32 |
| Несовпадающие участки | ||
| 1-12 | 1-19 | |
| 32-197 | 39-314 | |
| 200-229 | 317-347 | |
Длина выравнивания Mafft: 379
Cовпадающих колонок: ~20.7%* от Muscle
*Не учитывает одиночные совпадающие колонки!
| MUSCLE | T-Coffee | Длина |
|---|---|---|
| Совпадающие участки | ||
| 1-31 | 1-31 | 31 |
| 41-43 | 41-43 | 3 |
| 136-141 | 191-196 | 6 |
| 229-237 | 421-429 | 9 |
| 245-261 | 437-453 | 17 |
| Несовпадающие блоки | ||
| 32-40 | 32-40 | |
| 44-135 | 44-190 | |
| 142-228 | 197-420 | |
| 238-244 | 430-436 | |
Длина выравнивания T-Coffee: 453
Cовпадающих колонок: ~25.3%* от Muscle
*Не учитывает одиночные совпадающие колонки!
В результате попарного сравнения выравниваний, полученных с помощью программ Mafft и T-Coffee, с референсным выравниванием Muscle было установлено, что наибольшую степень сходства с референсом демонстрирует выравнивание, построенное T-Coffee. Несмотря на бóльшую протяжённость данного выравнивания, оно обладает бо́льшим количеством совпадающих с Muscle позиций. Выравнивание, полученное с помощью Mafft, несколько уступает T-Coffee по числу совпадающих блоков, однако различия между ними не являются критическими.
Сравнение результатов выравнивания по совмещению структур с выравниванием Muscle
В этом задании вновь пришлось обратиться к семейству белковых доменов из 11-го практикума. Были выбраны последовательности белков, содержащих этот домен (цепь А), далее представлены их PDB ID: 1g9x, 1ji0, 1xef, 1oxu. Также было выполнено два множественных выравнивания: первое — с помощью программы Muscle, второе — получено путём совмещения структур с использованием программы PDBeFold. Результат совмещения представлен на рисунке 1, для визуализации использовалась программа PyMOL. Кроме того, в Jalview был создан проект, содержащий оба выравнивания для их наглядного сравнения.
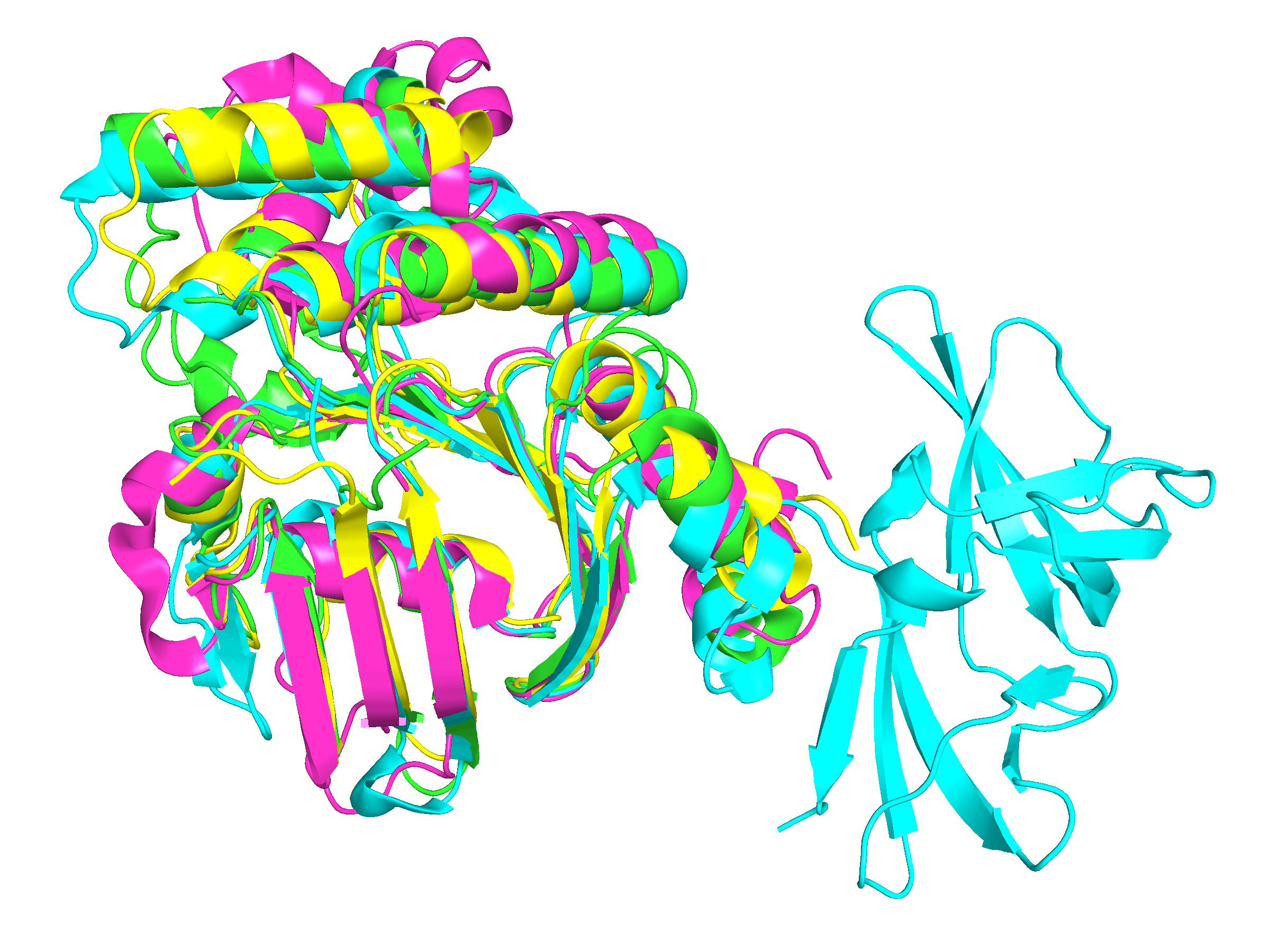
Рисунок 1. Совмещение 3D-структур белков
(1g9x:A – зеленый, 1ji0:A – желтый, 1xef:A – розовый, 1oxu:A – бирюзовый)
| MUSCLE | PDBeFold | Длина |
|---|---|---|
| Совпадающие блоки | ||
| 18-55 | 20-57 | 38 |
| 60-75 | 63-78 | 16 |
| 87-93 | 91-97 | 7 |
| 98-110 | 104-116 | 13 |
| 127-128 | 133-134 | 2 |
| 150-151 | 173-174 | 2 |
| 162-188 | 185-211 | 27 |
| 214-223 | 240-249 | 10 |
| 229-244 | 256-271 | 16 |
| 247-248 | 275-276 | 2 |
| 258-261 | 283-286 | 4 |
| 277-346 | 303-372 | 70 |
| 353-371 | 379-397 | 19 |
| Несовпадающие блоки | ||
| 1-17 | 1-19 | |
| 56-59 | 58-62 | |
| 76-86 | 79-90 | |
| 94-97 | 98-103 | |
| 111-126 | 117-132 | |
| 129-149 | 135-172 | |
| 152-161 | 175-184 | |
| 189-213 | 212-239 | |
| 224-228 | 250-255 | |
| 245-246 | 272-274 | |
| 249-257 | 277-282 | |
| 262-276 | 287-302 | |
| 347-352 | 373-378 | |
| 372-378 | 398-404 | |
Длина выравнивания Mafft: 404
Cовпадающих колонок: ~61.1%* от Muscle
*Не учитывает одиночные совпадающие колонки!
Проанализировав результаты сравнения выравнивания, полученного путём совмещения структур, с референсным выравниванием программы Muscle, можно отметить высокий уровень их сходства. Об этом свидетельствуют очень близкая длина выравниваний и большое количество довольно протяжённых совпадающих блоков.
Высокий процент совпадающих колонок между двумя выравниваниями также показывает, что программа Muscle в данном случае построила выравнивание, очень близкое к эволюционному. Его результат оказался схож с выравниванием по структурному совмещению, которое считается более правильным, поскольку структурные единицы эволюционно более устойчивы, чем первичная последовательность.
Программа MSA MAFFT
MAFFT (Multiple Alignment using Fast Fourier Transform) — это широко используемая высокопроизводительная программа для множественного выравнивания биологических последовательностей (MSA), разработанная Катохом и соавторами в 2002 году [1].
Основные подходы алгоритма MAFFT:
- Быстрое преобразование Фурье (FFT):
- Несколько стратегий выравнивания:
- Прогрессивные методы [2];
- Методы итеративного уточнения (значительно повышают точность за счёт многократного перевыравнивания) [3];
- Методы для больших данных [4].
- Добавление новых последовательностей:
Аминокислотная последовательность преобразуется в последовательность векторов, компонентами которых являются физико-химические свойства каждого остатка (объём и полярность по шкале Грантема). Корреляция между такими последовательностями вычисляется с помощью быстрого преобразования Фурье, что позволяет быстро находить гомологичные участки и снижает вычислительную сложность для консервативных участков последовательностей [1].
В MAFFT реализовано несколько подходов, которые можно настраивать в зависимости от размера данных и требуемой точности:
Со временем в программе появилась возможность добавлять новые последовательности (в том числе фрагментарные) в уже готовое выравнивание без его полной перестройки [2]. Это может быть особенно полезно при работе с данными секвенирований нового поколения.
Преимущества:
- MAFFT сочетает высокую скорость работы с точностью, сопоставимой или превосходящей такие программы, как ClustalW, T-Coffee и MUSCLE, особенно при работе с большими наборами данных [3].
- Программа использует нормализованную матрицу сходства (содержащую как положительные, так и отрицательные значения), что улучшает выравнивание последовательностей разной длины [1].
- MAFFT поддерживает многопоточные вычисления, что позволяет эффективно использовать многоядерные процессоры [2].
Недостатки:
- Несмотря на высокую скорость, методы итеративного уточнения могут требовать значительных вычислительных ресурсов при работе с очень большими наборами последовательностей.
- Для начинающих пользователей большое количество доступных подходов к выравниванию может затруднять выбор подходящего.
Доступность
MAFFT доступен как в виде командной строки, так и через веб-интерфейс, который предоставляет интерактивные инструменты для выбора последовательностей и визуализации результатов [4].
Список литературы
- Katoh K. et al. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform //Nucleic acids research. – 2002. – Т. 30. – №. 14. – С. 3059-3066.
- Katoh K., Standley D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability //Molecular biology and evolution. – 2013. – Т. 30. – №. 4. – С. 772-780.
- Katoh K. et al. MAFFT version 5: improvement in accuracy of multiple sequence alignment //Nucleic acids research. – 2005. – Т. 33. – №. 2. – С. 511-518.
- Katoh K., Rozewicki J., Yamada K. D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization //Briefings in bioinformatics. – 2019. – Т. 20. – №. 4. – С. 1160-1166.