| Главная | Семестры | Проекты | Обo мне | Ссылки | Заметки | Назад к оглавлению |
Парное выравнивание белков (вручную и с помощью Muscle)
- На данной странице вы можете увидеть, результаты моей работы по использованию скрипта perl и ознакомление/использование программы Jalview
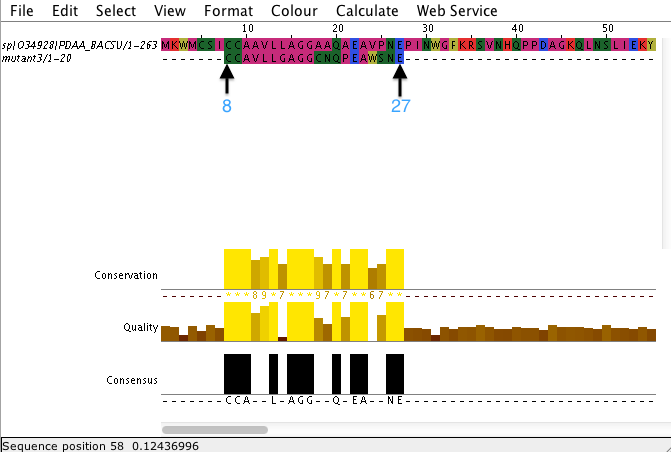
Выравнивание последовательности моего белка относительно искусственно синтезированных (с помощью скрипта с изменением параметров) коротких последовательностей
Скрипт моделирует мутант белка заданной длины (в а.о.) и с заданными параметрами. Параметры отвечают за вероятность изменения остатка (моделирующая "ошибку" ДНК-полимеразы) (-с), замены остатка при изменении позиции (-r), количество поколений (важное значение для анализирования изменений, например, в филогенетическом ряду) (-g), полноразмерные последовательности (-f), количество (-t) и длина пептида. Нажимая на номера выравниваний вы можете их посмотреть.
- Расцветка аминокислот:
- Алифатические - фиолетовым
- Ароматические - ораньжевым (светлозеленый)
- Отрицательно заряженные - синим
- Нейтральные гидрофильные - зеленым
- Положительно заряженные - красным
| Номер выравнивания | 1 | 2 | 3 |
Параметры:
|
|
|
|
| % идентичности | 30 | 39.1 | 60 |
| % сходства (по R) | 55 | 56.5 | 80 |
| вес по матрице BLOSUM62 | 19 | 31 | 57 |
{kind=link}
{kind=link}
{kind=link}
Из полученных данных мы можем сделать вывод о влиянии этих двух параметров на "эволюцию" наших пептидов. Параметр $def_change (моделирующий "ошибку" ДНК-полимеразы) отражает вероятность любого изменения в последовательности, а параметр $def_replace (замена остатка при изменении позиции) говорит о том, что с такой-то заданной вероятностью это изменение будет заключаться в замене. Таким образом, можно сравнить полученные результаты - удобнее сравнивать 1 и 3 относительно 2, т.к. и у 1 и у 3 только один из параметров не совпадает со вторым. Рассмотрим 1 и 2. У них вероятность изменения одинаковая, но во втором случае с большей вероятностью при изменении происходит замена (на 20%), по результатам видно, что в 2 случае проценты идентичности, сходства и вес по матрице BLOSUM62 существенно выше, что вполне очевидно, ведь в 2ом случае, если и происходит какая-то мутация, то с большей вероятностью она является заменой, т.е. точечна (тем более замена на идентичную а.к. произойдет с вероятностью 5% (1/20)), а в 1 она реже точечна, а чаще может являться делецией или инсерцией, а значит выравнивание будет смещаться (эта вставленная/вырезанная часть последовательности не будет состыковыватся с той, по которой мы выравниваем). Сравнивая 2 и 3, можно придерживаться близкой логики, ведь в данном случае изменяется вероятность любой замены (в 3ем замена случается на 20% реже) => у нас меньше проблем, что последовательность изменилась.
Выравнивание последовательности моего белка c его гомологами /ортологов
Выравнивание осуществляется не вручную, а с помощью встроенной возможности Jalview 2.8 (Web Service -> Alignment -> Muscle with default). Затем с помощью программы infoalign из пакета EMBOSS находится нужная информация о каждой паре выравниваний (см. ниже).
Описание программы infoalign из пакета EMBOSS:
Программа выводит данные о множественном (и о парном) выравнивании последовательностей.
Ее важные опции:
- idcount показывает количество одинаковых позиций
- simcount показывает число похожих позиций
- difcount показывает число различных позиций
- gaps показывает количество пропусков
- gapcount показывает число аминокислот напротив пропуска (gap)
Информация о выравниваниях:
- PDAA_BACSU и PDAB_BACSU
Name Sequence Length Aligned Length Gaps Gap Length Identity Similarity Difference % Change Weight PDAA_BACSU/1-263 263 278 4 15 263 0 0 5.395683 1.000000 PDAB_BACSU/1-254 254 278 3 24 85 43 126 69.424461 1.000000 - PDAA_BACSU и B9IXV4_BACCQ
Name Sequence Length Aligned Length Gaps Gap Length Identity Similarity Difference % Change Weight PDAA_BACSU/1-263 263 298 4 35 263 0 0 11.744967 1.000000 B9IXV4_BACCQ/1-275 275 295 4 20 116 52 107 60.677967 1.000000 - PDAB_BACSU и B9IXV4_BACCQ
Name Sequence Length Aligned Length Gaps Gap Length Identity Similarity Difference % Change Weight PDAB_BACSU/1-254 254 280 4 26 254 0 0 9.285714 1.000000 B9IXV4_BACCQ/1-275 275 276 1 1 100 54 121 63.768116 1.000000
Если интересно, вы можете посмотреть выравнивание всех трех гомологов ниже. Они окрашены по встроенной цветовой схеме Clustalx с заданным мной порогом идентичности равным 67% (чтобы окрашивание происходило, когда хотя бы 2 из 3 аминокислот находятся на одном уровне, они окрашивались в соответствующие природе элемента цвета).
.jpg)
Выравнивание последовательности моего белка относительно искусственно синтезированных (с помощью скрипта evolve_protein.pl, в котором можно изменять параметры) последовательностей (длиной 263 как и у моего белка). Значимо то, что эти параметры эколюционирования близки к реальным.
| Номер выравнивания | 1 | 2 | 3 |
Параметры:
|
|
|
|
| % идентичности | (263-21)/263*100%=~92.02% | (263-181)/263*100%=~31.18% | (263-22)/263*100%=~91.63% |
Как мы видим, при вероятности изменения остатка (параметр отвечающий за моделирование "ошибки" ДНК-полимеразы) равной 0.0001 и большом количестве поколений - 10000 (2 "мутант") процент идентичности резко падает в отличии от тех вариантов, когда либо меньше поколений (1), либо вероятность изменения остатка меньше (3). Эти результаты вполне логичны, ведь при большем "ошибании" ДНК-полимереразы больше будет изменений и чем больше поколений пройдет чем больше у них будет "ошибок" предковых (которые были созданы при создании предыдущих поколений) и своих (которые произошли при создании их). Идентичность у 1 и 3 "мутанта" почти совпадает, ведь в одном случае у нас была большая "ошибка" полимеразы (в 10 раз), но зато мешьше поколений (тоже в 10 раз), а в другом "ошибка" была меньше, но зато поколений больше, причем эта разница между количеством поколений и процентом "ошибок" сопоставима - отсюда и такие результаты. Но как мы видим по этим результатам (1 и 3) вероятность "ошибки" ДНК-полимеразы вносит немного больший вклад в изменчивость, чем количество поколений.
На создание 1000 поколений E.Coli при делении клетки раз в 30 минут ей потребуется ~9.9658*30=298.97мин=4ч 59мин, так как увеличение количества поколений будет происходить как 2^(количество делений (в начальный момент она не поделилась, а значит 0 => 1 E.Coli)) => количество делений=log2(1000) и количество делений мы уножаем на 30 мин, так как 1 деление каждые 30 мин происходит. А на создание 10000 - 13.2877*30=398.63мин=6ч 39мин.