| Главная | Семестры | Проекты | Обo мне | Ссылки | Заметки | Назад к оглавлению |
BLAST
- BLAST (англ. Basic Local Alignment Search Tool) — семейство компьютерных программ, служащих для поиска гомологов белков или нуклеиновых кислот, для которых известна первичная структура (последовательность) или её фрагмент. Используя BLAST, исследователь может сравнить имеющуюся у него последовательность с последовательностями из базы данных и найти последовательности предполагаемых гомологов. Является важнейшим инструментом для молекулярных биологов, биоинформатиков, систематиков.
Результаты поиска гипотетических гомологов белка PDAA_BACSU:
| Поиск по Swiss-Prot | Поиск по PDB | Поиск по "nr" | |
| 1. Лучшая находка (с последовательностью исходного белка) | |||
| Accession | O34928.1 | 1W17_A | YP_006628965.1 |
| E-value | 0.0 | 0.0 | 0.0 |
| Вес (в битах) | 554 bits | 554 bits | 554 bits |
| Процент идентичности | 100% | 100% | 100% |
| 2. Число находок с E-value < 10–10 | 1 | 3 | 13 |
| 3. "Худшая из удовлетворительных" находка (последняя в выдаче с E-value < 1) | |||
| Номер находки в списке описаний | 27 | 15 | 100 |
| Accession | O13842.1 | 4HD5_A | ZP_17375824.1 |
| E-value | 0.004 | 0.76 | 5e-92 |
| Вес (в битах) | 41.2 bits | 31.2 bits | 283 bits |
| % идентичности | 19/63(30%) | 32/106(30%) | 131/252(52%) |
| % сходства | 36/63(57%) | 48/106(45%) | 180/252(71%) |
| Длина выравнивания | 63 | 106 | 252 |
| Координаты выравнивания (от-до, в запросе и в находке) | 63-125;66-127 | 62-156;194-294 | 2-253;4-255 |
| Число гэпов | 1/63(1%) | 16/106(15%) | 0/252(0%) |
Анализирование результатов поиска PDAA_BACSU по разным БД в BLASTP:
- Удалось ли найти исходный белок в Swiss-Prot и "nr", а его структуру в PDB? Да
- Сравнение числа явных гомологов (E-value < 1e-10) при поиске по разным БД и пояснение возможных причин различий. Для Swiss-Prot 20, PDB 10, nr 100. Банк nr включает в себя последовательности из всевозможных источников для программы BLAST и следовательно в нем огромное количество последовательностей белков, в Swiss-Prot меньше последовательностей, тк это одна база данных, в которую к тому же попадают только "проверенные" последовательности (подробнее здесь). В БД PDB и того меньше последовательностей, ведь чтобы они там оказались их третичную (или + четвертичную) структуры надо проанализировать.
- Сколько всего находок и каков E-value самой последней находки? Чем в вашем случае было лимитировано число находок: значением E-value или заданным по умолчанию предельным размером выдачи?В Swiss-Prot, PDB в последних находках значение E-value 8.7 и 8.5 соответственно, а размер выдачи меньше 100 (а это максимальное значение выдачи по умолчанию) в обоих случаях и следовательно тут выдача была ограничена значением E-value (он по умолчанию 10) .В nr: всего находок 100, E-value последней находки 5e-92 (сильно меньше максимального, т.е. 10), количество находок было ограничено заданным по умолчанию предельны размером выдачи.
Поиск гипотетических гомологов изучаемого белка с фильтром по таксонам. Искать буду в "nr", так как там самый большой выбор результатов.
Хочется найти гомологи как можно более близкие по последовательности, но при этом как можно более удаленные филогенетически. Лучшие находки по таксонам:
Царство Eukaryota
- Номер находки в списке описаний: 1
- Accession: EGF76050.1
- E-value: 1e-24
- Вес (в битах): 107 bits
- % идентичности: 71/249(29%)
- % сходства: 127/249(51%)
- Длина выравнивания: 249
- Координаты выравнивания (от-до, в запросе и в находке): 14-253; 33-257
- Число гэпов: 33
Отдел Actinobacteria
- Номер находки в списке описаний: 1
- Accession: ZP_10070839.1
- E-value: 2e-33
- Вес (в битах): 128 bits
- % идентичности: 74/192(39%)
- % сходства: 109/192(56%)
- Длина выравнивания: 192
- Координаты выравнивания (от-до, в запросе и в находке): 64-253; 54-239
- Число гэпов: 8
Класс Clostridia
- Номер находки в списке описаний: 1
- Accession: YP_001211023.1
- E-value: 1e-81
- Вес (в битах): 255 bits
- % идентичности: 117/235(50%)
- % сходства: 159/235(67%)
- Длина выравнивания: 235
- Координаты выравнивания (от-до, в запросе и в находке): 19-253; 82-315
- Число гэпов: 1
Порядок Lactobacillales
- Номер находки в списке описаний: 3
- Accession: YP_004374799.1
- E-value: 2e-36
- Вес (в битах): 139 bits
- % идентичности: 75/206(36%)
- % сходства: 112/206(54%)
- Длина выравнивания: 206
- Координаты выравнивания (от-до, в запросе и в находке): 61-262; 37-238
- Число гэпов: 8
Семейство Listeriaceae
- Номер находки в списке описаний: 8
- Accession: ZP_00230265.1
- E-value: 1e-33
- Вес (в битах): 128 bits
- % идентичности: 74/191(39%)
- % сходства: 105/191(54%)
- Длина выравнивания: 191
- Координаты выравнивания (от-до, в запросе и в находке): 66-256; 221-404
- Число гэпов: 7
Род Geobacillus
- Номер находки в списке описаний: 3
- Accession: YP_003252435.1
- E-value: 4e-116
- Вес (в битах): 337 bits
- % идентичности: 155/263(59%)
- % сходства: 202/263(76%)
- Длина выравнивания: 263
- Координаты выравнивания (от-до, в запросе и в находке): 1-263; 9-268
- Число гэпов: 3
Вид Bacillus anthracis
- Номер находки в списке описаний: 3
- Accession: 2J13_A
- E-value: 4e-95
- Вес (в битах): 282 bits
- % идентичности: 131/231(57%)
- % сходства: 173/231(74%)
- Длина выравнивания: 231
- Координаты выравнивания (от-до, в запросе и в находке): 23-253; 12-242
- Число гэпов: 0
Мне больше всего нравится результат по поиску в отделе Actinobacteria, так как при такой филогенетической удаленности организмов очень любопытно найти организмы с такими показателями, в частности идентичности последовательности - 39%! Как мы видим далее с порядком Lactobacillales лучший результат и то хуже, хотя группы организмов более поздно разошлись на филогенетическом древе.
BLAST двух последовательностей:
С помощью программы BLAST создается парное выравнивание белка pdaa_bacsu и его гомолога pdab_bacsu. Для охарактеризования выравнивания используются карты локального сходства. Можете посмотреть на них в двух видах: с порогом на E-value, равным 10 (по умолчанию) и с порогом на E-value, равным 0.01.
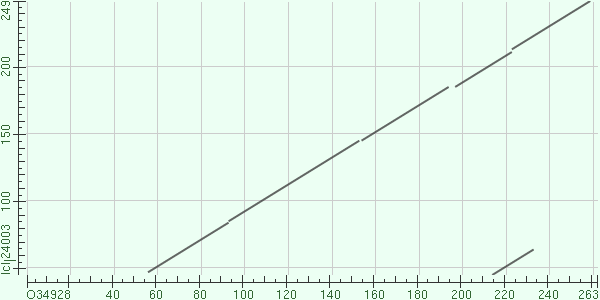
|
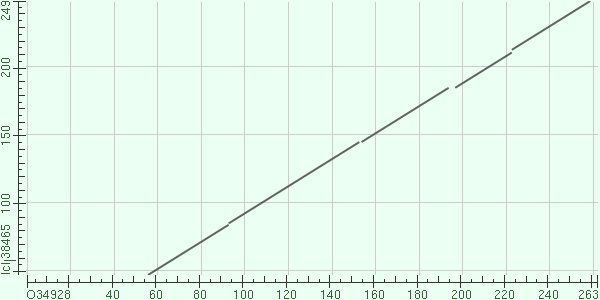
|
| Рис. 1. Карта локального сходства с порогом E-value равным 10. | Рис. 2. Карта локального сходства с порогом E-value равным 0.01. |
Графики получились различными для разных значений параметра E-value. Это случилось потому, что для маленького участка сходства (рис. 1) значение E-value больше 0.01, а большой участок совпал на первом и втором рисунке.
Сравнение результатов поиска с различными матрицами BLOSUM
Сравним результаты матрицы BLOSUM62 и BLOSUM45 по лучшему (с моей точки зрения) удаленному филогенетически гомологу, а им являлся результат по запросу таксона: отдел Actinobacteria. Как упоминалось ранее в другой моей работе (предыдущей: "Применение алгоритмов парных выравниваний к белку PDAA_BACSU") число после названия матрицы обозначает порог кластеризации.
Для матрицы BLOSUM62 результаты выше (не вижу смысла их повторять на одной странице несколько раз). А для матрицы BLOSUM45 смотрите ниже:
Отдел Actinobacteria
- Номер находки в списке описаний: 1
- Accession: ZP_10070839.1
- E-value: 2e-33
- Вес (в битах): 128 bits
- % идентичности: 74/192(39%)
- % сходства: 109/192(56%)
- Длина выравнивания: 192
- Координаты выравнивания (от-до, в запросе и в находке): 64-253; 54-239
- Число гэпов: 8
Следовательно результаты совпали.
Сравнение различных интерфейсов программы BLAST
Программой BLAST можно пользоваться на различных серверах, таких как NCBI (которым пользовалась я для выполнения вышеизложенных иследований), EMBL-EBI и UniProt.
В интерфейсе EMBL-EBI мне не понравилось, что нельзя вводить accession number и gi, а только непосредственно последовательность вручную или с помощью загрузки файла - это не удобно. В UniProt же мне не понравилось, что нужно при прочих равных (количество находок в том и другом случае выставлено 100) ждать относительно долго - ~30сек, а в EMBL-EBI и NCBI около 12. В целом мне больше понравился интерфейс у NCBI, хотя, стоит заметить, что у EMBL-EBI есть значительный плюс, он состоит в том, что когда смотришь результаты, они представлены более компакно и можно по выбору отмечать те, результаты, которые хочется посмотреть с выравниваниями и те что без (с помощью отметки галочкой необходимого и задачи нужного слева) и еще (!) выравнивания можно скачать в fasta формате. Единственное достоинство, которое мне импонировало на данный момент в UniProt BLAST, заключается в том, что есть колонка с "звездочками" в результатах, которая показывает какие последовательности лежат в Swissprot, а какие в TrEMBL (подробнее об разнице между ними вы можете прочитать в моей работе "UNIPROT").