| Главная | Семестры | Проекты | Обo мне | Ссылки | Заметки | Назад к оглавлению |
Комплексы ДНК-белок
Задание 1. Поиск ДНК-белковых контактов в заданной структуре
Упр.1 Вспомним, как с помощью команды define JMol задавать множества атомов:
- множество атомов кислорода 2'-дезоксирибозы(set1)
- множество атомов кислорода в остатке фосфорной кислоты (set2)
- множество атомов азота в азотистых основаниях (set3)
Скрипт можно посмотреть тут.
Упр.2 Опишем ДНК-белковые контакты в структуре 1MHD.pdb. Сравниим количество контактов разной природы.
Будем считать полярными атомы кислорода и азота, а неполярными атомы углерода, фосфора и серы. Назовем полярным контактом ситуацию, в которой расстояние между полярным атомом белка и полярным атомом ДНК меньше 3.5 А. Аналогично, неполярным контактом будем считать пару неполярных атомов на расстоянии 4.5 А. Найденные атомы можно увидеть на рисунке 1. Результаты приведены в таблице:
| Контакты атомов белка с | Полярные | Неполярные | Всего |
| остатками 2'-дезоксирибозы | 1 | 9 | 10 |
| остатками фосфорной кислоты | 8 | 0 | 8 |
| остатками азотистых оснований со стороны большой бороздки | 2 | 8 | 10 |
| остатками азотистых оснований со стороны малой бороздки | 0 | 3 | 3 |
 |
| Рис. 1. |
Упр.3
Получим изображение (рис. 2 и 3) с помощью команды:
nucplot 1MHD_old.pdb (1MHD_old.pdb уже получен с помощью команды: remediator --old "1MHD.pdb" > "1MHD_old.pdb")
 |
 |
| Рис. 2-3. Схема ДНК-белковых контактов полученная с помощью программы nucplot. |
Упр.4. На полученной схеме выберем:
- аминокислотный остаток с наибольшим числом указанных на схеме контактов с ДНК, таким является Arg74, он связан с G2004 NH2 и NH1 (рис. 4)
- аминокислотный остаток наиболее важный для распознавания последовательности ДНК - Глутамин 76 (он наиболее важен по моему мнению, потому что на схеме 2 показана связь не с сахарофосфатным остовом, а с самим азотистым основанием, и, как видно на рис. 5, глутамин четко лежит в бороздке ДНК, что свидетельствует о том, что это контакт высоко специфичен).
 |
| Рис. 4. Контакт Gly74 c G2004. |
 |
 |
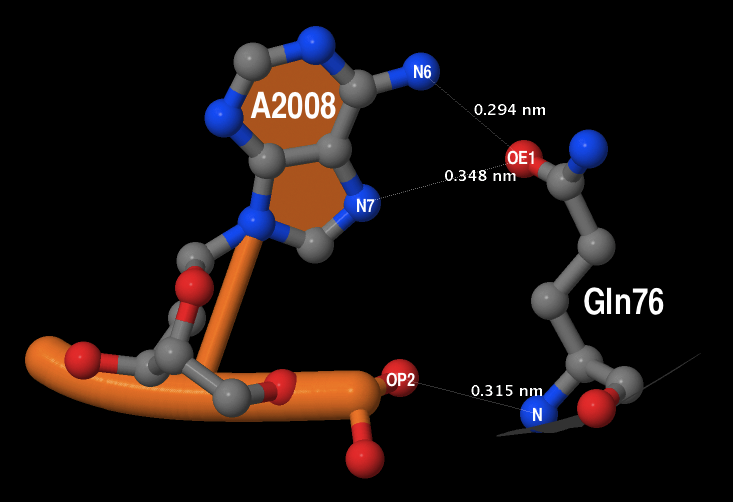 |
| Рис. 5. Контакт Gln76 с A2008. |
Задание 2. Предсказание вторичной структуры заданной тРНК
Упр.1 Предсказание вторичной структуры тРНК путем поиска инвертированных повторов:
Программа einverted из пакета EMBOSS позволяет найти инвертированные участки в нуклеотидных последовательностях. Найдем возможные комплементарные участки в последовательности исследуемой тРНК. Сравним с их описанием, полученным ранее с помощью find_pair. Результаты сравнения можно посмотреть в таблице, приведенной ниже. Постараемся подобрать параметры для получения предсказания, наиболее близкого к реальной структуре.
Пользуемся командой:
einverted 1H4S.fasta
Использованные параметры и полученный файл:
- Gap penalty: 12
- Minimum score threshold: 0
- Match score: 3
- Mismatch score: -2
- Файл: seq1.inv
Упр.2 Предсказание вторичной структуры тРНК по алгоритму Зукера.
Программа mfold из пакета EMBOSS реализует алгоритм Зукера. Получим 6 предполагаемых структуру из них выберем 1 наиболее близкую к реальной (Рис. 6).
| |
| Рис. 6. |
Структуру РНК из JMol можно увидеть на рис. 7-9.
 |
 |
 |
| Рис. 7-9. 1H4S тРНК. Акцепторый стебель окрашен белым, D-стебель - зеленым, T-стебель - желтым, антикодоновый - синим, а антикодоновый триплет оранжевым. |
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель (white) | (5')4-7(3') (5')66-69(3') 4 пары |
предсказано 0 пар
|
предсказано 0 пар (смещение на 1 основание)
|
| D-стебель(green) |
(5')10-13(3')
(5')22-25(3')
4 пары
|
предсказано 0 пар
|
предсказано 0 пар (смещение на 1 основание)
|
| T-стебель (yellow) |
(5')49-54(3')
(5')58,61-65(3')
6 пар
|
предсказано 0 пар
|
предсказано 0 пар
|
| Антикодоновый стебель (blue) |
(5')28-34(3')
(5')38-44(3')
7 пар
|
предсказано 0 пар
|
предсказаны все 7 пар!
|
Общее число канонических пар нуклеотидов |
23
|
19
|
21
|