| Главная | Семестры | Проекты | Обo мне | Ссылки | Заметки | Назад к оглавлению |
Сравнение расшифрованной структуры в ЯМР и РСА
Выбор структуры
Для анализа был выбран белок (ацетил-СоА связывающий белок быка), имеющий ЯМР и РСА структуры. ЯМР структура: 1ACA, эта запись содержит 20 моделей (рис. 1). А РСА структура: 1HB6 и ее разрешение 2 А (рис. 2).
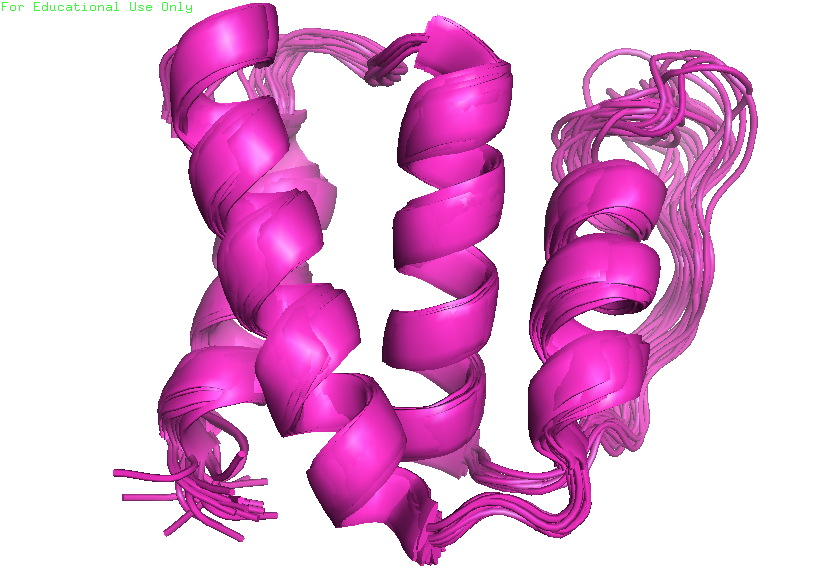 |
| Рис. 1. ЯМР структура белка. |
 |
| Рис. 2. Структура белка, полученная РСА. |
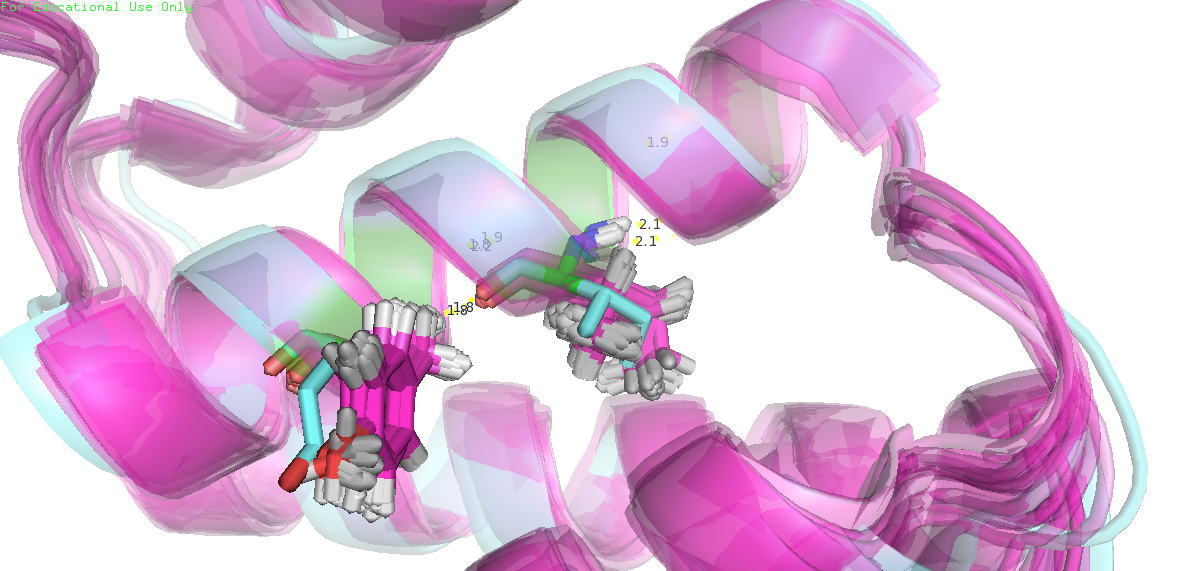
Чтобы понять насколько структура в растворе совпадает с структурой в кристале выравнили их не гибко с помощью сервера POSA (сервер гибкого множественного выравнивания) и в PyMol с помощью команды align 1aca, 1hb6. Получили одно и тоже (рис. 3). Видео можете скачать тут
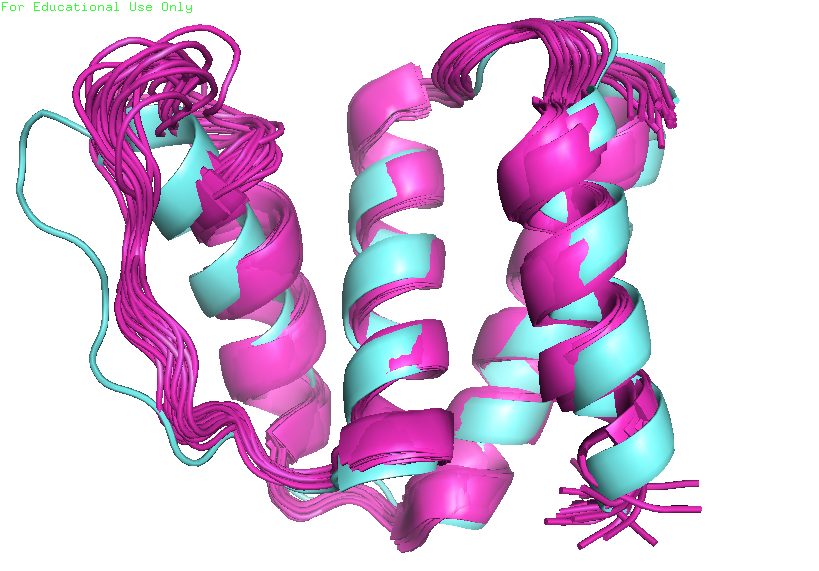 |
| Рис. 3. Пространственное выравнивание 1ACA (ЯМР; розовый) и (РСА; голубой). |
Анализ водородных связей
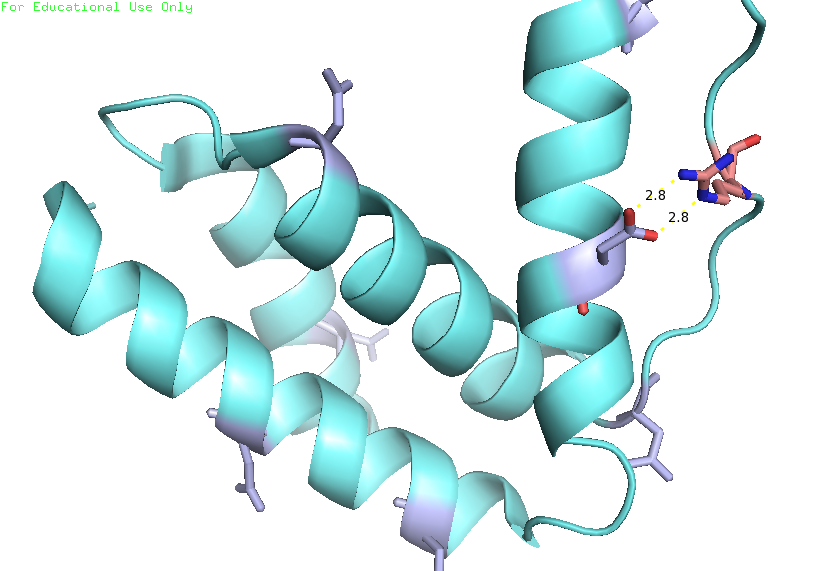
1. Проанализируем водородные связи в петле на поверхности глобулы. На рисунке 4 можно увидеть пример такой водородной связи.
 |
 |
| Рис. 4. Водородная связь между аргинином (серый) и аспартатом (розовый). Длина водородных связей в РСА структуре 2.8 А. Все модели, полученные методом ЯМР, имеют эти две водородные связи. Минимальная длина составляет 2.2 А, а максимальная 3.2 А. Медианная получилась равна 2.6 А. |
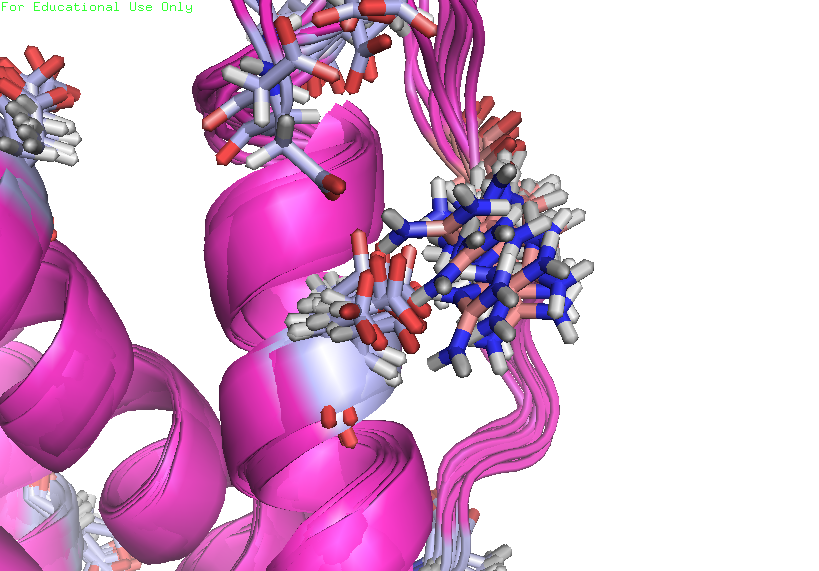
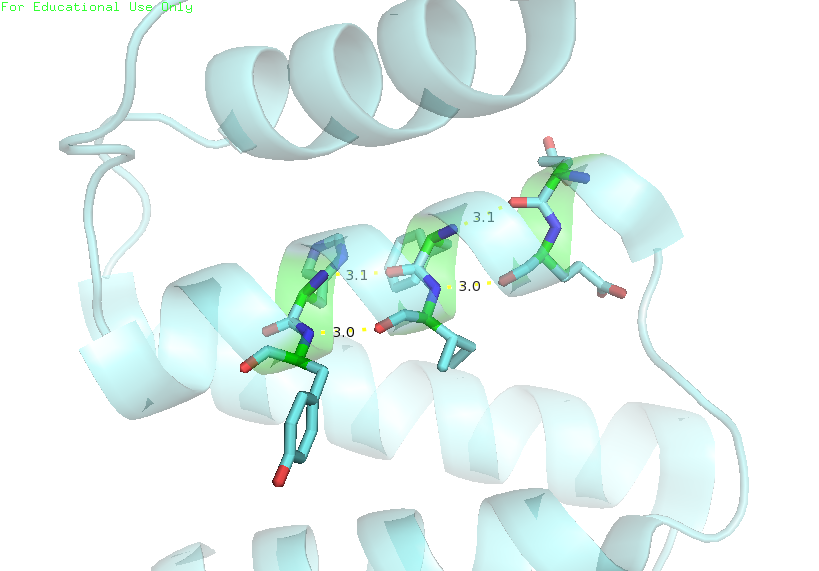
2. Проанализируем водородные связи, находящиеся в остове, в альфа-спиралях. На рисунке 5 можно увидеть примеры таких водородных связей.
 |
 |
| Рис. 5. Остовные водородные связи в альфа спирали. С-альфа атомы покрашены в зеленый цвет, синие азоты и красные кислороды. Длина водородной связи в структуре РСА 3 А. Все модели, полученные методом ЯМР, имеют все эти водородные связи, для дальнейшего анализа была взята водородная связь между ILE27 и TYR31. Ее минимальная длина составляет 2.1 А, а максимальная 2.8 А. Медианная получилась равна 2.4 А. |
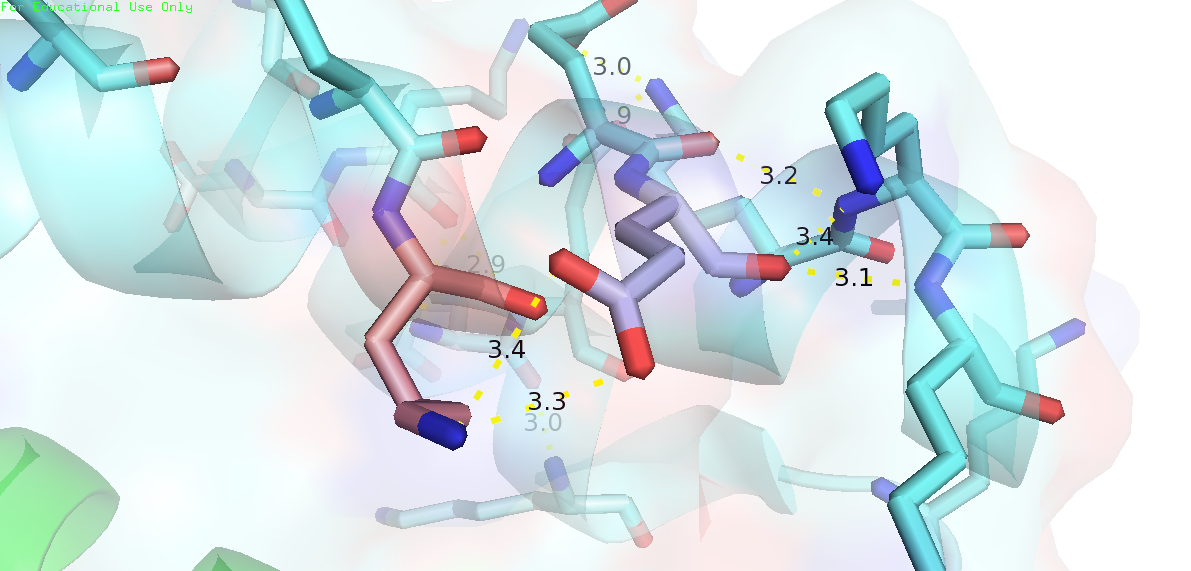
3. Проанализируем водородную связь боковых цепей в ядре белка. На рисунке 6 можно увидеть пример такой водородной связи.
|
| Рис. 6. Водородная связь между радикалами глутаминовой кислотой (серый) и лизином (розовый), которые находятся в одной альфа-спирали. Длина водородной связи в структуре РСА 3.3 А. Все модели, полученные методом ЯМР, имеют эту водородную связь. Ее минимальная длина составляет 2.7 А, а максимальная 3.3 А. Медианная получилась равна 2.9 А. |
Сводная таблица с информацией о водородных связях:
| Расположение Н-связи. Донор и акцептор протонов | Длина водородной связи в РСА | ЯМР | |||
| Число моделей имеющих Н-связь | Наименьшая длина Н-связи, A | Наибольшая длина Н-связи, A | Медианная длина Н-связи, A | ||
| между радикалами, в петле, выходящей на поверхность глобулы; акцептор: Asp, донор: Arg | 2.8 (обе) | 20/20 | 2.2 | 3.2 | 2.6 |
| между остовами в ядре белка, формирует альфа-спираль; акцептор: Ile, донор: Tyr | 3.0 | 20/20 | 2.1 | 2.8 | 2.4 |
| между боковыми цепями в ядре белка, акцептор: Glu, донор: Lys | 3.3 | 20/20 | 2.7 | 3.3 | 2.9 |
Из всего вышесказанного и наблюдаемого можно сделать выводы, что белок имеет очень похожие структуры в кристалле и белке, что длина одной и той же водородной связи в модели ЯМР будет обычно короче, чем в РСА из-за того, что ЯМР распознает водороды, а РСА почти всегда нет.
Совместим нашу РСА структуру с моделями ЯМР с помощью сервера PDBeFold. Структуры отлично выравниваются, можно посмотреть выдаваемый итоговый отчет:
## Q-score P-score Z-score RMSD Nalgn Nsse Ngaps Seq-% Nmd Nres-Q Nsse-Q Nres-T Nsse-T Query Target
1 0.6858 4.63 6.218 1.672 82 4 2 0.9878 0 87 4 86 4 PDB 1aca:A PDB 1hb6:A
Некоторые из используемых команд:
#чтобы сделать видео выравнивания mset 1,180 util.mroll 1, 180, 1 set ray_trace_frames, 1 set cache_frames, 0 #чтобы увидеть все модели ямр структуры сразу split_state 1aca #анализ водородных связей remove solvent show surface set transparency, 0.5 show sticks, Asp or R distance hbonds, Asp, R, 3.5, mode=2 distance hbonds, sele, sele, 3.5, mode=2 sel sele and /////ca set label_size, -0.1