| Главная | Семестры | Проекты | Обo мне | Ссылки | Заметки | Назад к оглавлению |
Анализ трехмерных структур
Определение вторичной структуры
Задание 1. Определить вторичную структуру одного из белков.
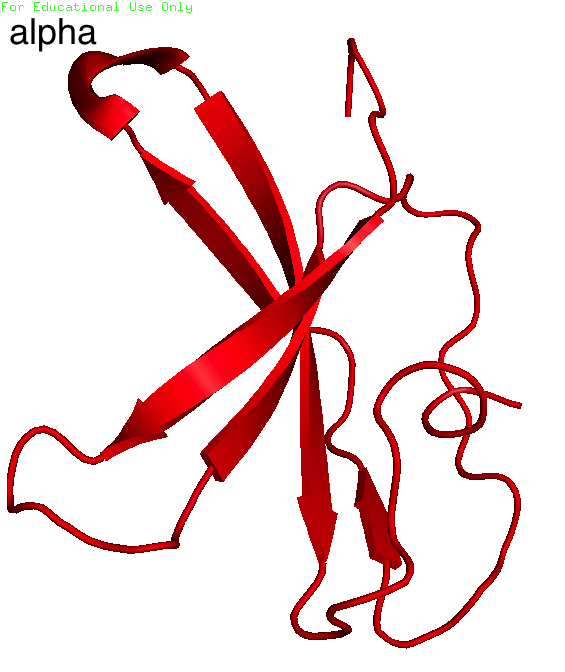
Для анализа была выбрала структура индуцируемого гипоксией фактора 1 альфа - 3hqu (рис. 1). Видео можно посмотреть тут.
Определим вторичную структуру с помощью программы DSSP.Stride не дал результатов. Сервер DSSP и Stride.
{kind=link}
 |
| Рис. 1. Структура индуцируемого гипоксией фактора 1 альфа - 3hqu. |
Таблица сравнения элементов вторичной структуры:
| Элемент | Границы в DSSP | Границы в PDB-файле |
| α-спираль | 190-193 | 190-196 |
| α-спираль | 199-205 | 198-210 |
| α-спираль | 216-231 | 216-231 |
| β-тяж | 240-242 | 240-242 |
| 310-спираль | 247-249 | 247-249 |
| β-тяж | 251-252 | 251-252 |
| β-тяж | 255-259 | 255-259 |
| α-спираль | 267-282 | 267-282 |
| β-тяж | 292-295 | 292-295 |
| β-тяж | 298-303 | 298-303 |
| β-тяж | 308-313 | 308-313 |
| β-тяж | 322-329 | 322-329 |
| α-спираль | 336-339 | 336-339 |
| β-тяж | 343-345 | 343-345 |
| β-тяж | 354-356 | 354-356 |
| β-тяж | 362-367 | 362-367 |
| β-тяж | 374-379 | 374-379 |
| β-тяж | 383-392 | 383-392 |
| α-спираль | 393-398 | 393-398 |
Красные в таблице несовпадающие участки. В целом сервер DSSP очень хорошо определили вторичную структуру белка, из таблицы видно, что несовпали только границы двух α-спиралей, находящихся в начале структуры. Сервер сделал их короче, чем в PDB файле.
Интересным элементом в данной структуре является "перекрученная" 310 спираль. Она встретилась на учатке петли в повороте между β-тяжами.
Задание 2. С помощью SheeP построить карту одного из бета-листов в выбранной вами структуре белка и сопоставить с изображением этого листа.
Получили карту β-листа с помощью сервера SheeP (рис. 2). Каждый столбец карты соответствует одному хребту (crest) β-листа. Всего программа обнаружила 3 β-листа, что верно (рис. 1). Для построения хребта был выбран 7 столбец (рис. 2, синий). Для данного β-листа нельзя сделать выводов о том, какие из остатков (на рис. 2 на желтом или красном фоне) обращены в сторону гидрофобного ядра молекулы, так как этот лист находится в центре белковой глобулы. Зато можно сделать вывод, что красные аминокислотные остатки обращены в сторону спирали, а желтые в противоположную - к другому β-листу. Для построения схемы водородных связей (3 тяжа, 4-5 гребней) участок выделенный оранжевым (рис. 3).
 |
|
 |
 |
| Рис. 2. Сверху: один из β-листов, определенных SheeP. Снизу: он же в PyMol (розовый). В виде сфер выделены Cα-атомы, принадлежащие выбранному хребту (выделенный синим столбец). |
 |
| Рис. 3. Схема водородных связей оранжевого участка. |
Команды для создания схемы водородных связей:
sel hres, i. 384-388 or i. 298-302 or i. 255-259 distance hbonds, hres, hres, 3.5, mode=2 sel hres and (/////ca or /////c or /////n or /////o) show sticks, sele set dash_color, brown
Совмещение структур и структурных гомологов
Задание 1. Построить совмещение структур белка с его четырьмя структурными гомологами.
Для анализа была взята структура 1ACA (из задания по ЯМР). Поиск гомологов осуществляем с помощью PDBeFold. Из результата были выбраны 4: 3t49:C, 2pmr:A, 4wwi:A, 2m5a:A. и совмещены (рис. 4). Выравнивание гомологов по структуре можно увидеть (в JalView; окраска Сlustalx) (рис. 5 вверху). Далее выровним последовательности в программе JalView с помощью алгоритма Muscle (рис. 5 внизу). Проект с двумя этими выравниваниями можно скачать тут.
 |
| Рис. 4. Совмещение структуры 1aca и 4 ее структурных гомологов. |
 |
| Рис. 5. Выравнивание по структуре и по последовательности (алгоритмом Muscle) гомологов 1aca (в JalView; окраска Сlustalx). |
Если сравнить множественное выравнивание по последовательности с выравниванием по структуре, можно заметить, что они совсем не похожи, даже длина у них разная. Все потому, что много гэпов и за них штафуют. Наверно взяла слишком разные последовательности. Попробуем другие: 2fdq:B, 4fzo:B, 2ahm:A, 2pmr:A (рис. 6). Сделаем тоже самое (рис. 7).
 |
| Рис. 6. Совмещение структуры 1aca и 4 ее еще одних структурных гомологов. |
 |
| Рис. 7. Выравнивание по структуре и по последовательности (алгоритмом Muscle) гомологов 1aca (в JalView; окраска Сlustalx). |
Все равно получились не сильно похожие выравнивания. Возьмем довольно интересное отличие (выделено на рисунке 7 фиолетовым). Тут в структурном выравнивании с лизинами выровнилась глутаминовая кислота, тогда как при выравнивании последовательностей с этими же лизинами выровнилась другая положительно заряженная кислота - аргинин. Если рассуждать с точки зрения зарядов, то выравнивание по последовательностям смотрится более верным. Но, если мы посмотрим на структурное выравнивание (рис. 8), становится понятно, что глутаминовая кислота тут лежит между двумя большими альфа-спиралями, тогда как аргинин находится в альфа-спирале. Таким образом, структурное выравнивание в данном случае правильнее.
 |
| Рис. 8. Структурное выравнивание. Синим выделена глутаминовая кислота, красным - аргинин. |
Задание 2. Использовать поиск по сходству структур в PDBeFold.
Для задания выбран домен 1b0m A:203-315. Найдем сходные с ним структуры (рис. 9). Отсортируем результаты по RMSD. Выдачу можно посмотреть тут. Получилось 37 результата, среди которых нет исходного домена. Разберемся в чем дело. Попробуем найти этот домен в полной структуре 1b0m. Получили 16% общих вторичных структур (процент sse), вспомним, что при первом поиске мы использовали 70% минимального сходства (см. рис. 9), следовательно, если установим это значение равным 15%, то получим в результатах исходный домен. Попробуем. Действительно, теперь мы получили исходный домен в результатах (можно посмотреть 4660 результата).
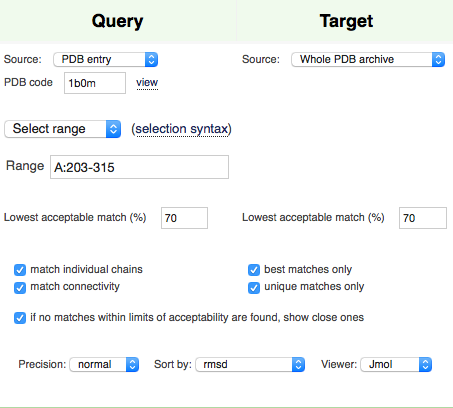 |
| Рис. 9. Запрос для поиска по сходству структур домена 1b0m A:203-315 в PDBeFold. |
Задание 3. Совместить по заданому выравниванию.
Для выполнения задания выбрала структуру 2f53. Вы можете увидеть одну структуру константного домена T-клеточного рецептора из цепочки α (d:115-191) и одну из - β (e:113-241) на рисунке 10. Далее построили карты β-листов с помощью SheeP в одной ориентации (flip columns) (рис. 11 и 12).
 |
 |
| Рис. 10. Домены человеческого Т-клеточного рецептора из α и β цепочки. |
 |
| Рис. 11. Карта β-листа цепочки α. |
 |
| Рис. 12. Карта β-листов цепочки β. |
Теперь построим выравнивание этих β-листов, чтобы консервативные цистеины и остатки, спаренные с ними, оказались выровнеными (рис. 13). Команда в PyMol:
pair_fit A///121-125+134-139+174-179+155-157/CA, B///121-125+140-145+186-191+167-169/CA
 |
| Рис. 13. Выравнивание β-листов с помощью PyMol. |
Как видно на рисунке 13, в этом совмещение нерегулярные петли имеют одинаковую топологию, хотя и на месте некоторых участков нерегулярной структуры α у β цепочки находятся регулярные элементы (спирали и тяжи).
Нахождение гидрофобных кластеров
Задание 1. Найти гидрофобные кластеры в структуре какого-либо белка.
Используя Clud найдем гидрофобные кластеры в структуре 1VPW.
| Параметры кластера | Найденные кластеры, количество остатков | Иллюстрация | Пояснение к результату |
| 5.4 А, минимальное число атомов 3 (параметры по умолчанию) | 268,244,60,20,14,7,6,2по4,5по3 |  |
можно заметить, что наиболее большие кластеры получились в областях наибольшей плотности регулярных структур; а более мелкие кластеры поддерживают неохваченные элементы вторичной структуры |
| 4.5 А, минимальное число атомов 15 | 80,64,58,46,41,22,19,17 |  |
"ужестили" параметры; наши большие кластеры разбились на поменьше; тепрь кластеры в областях наибольшей плотности альфа-спиралей и отдельно бета-листов |
| 3.5 А, минимальное число атомов 3 | 6,5,4по4,5по3 |  |
слишком жесткие параметры; остались какие-то кусочки кластеров |
На первом рисунке в таблице выше есть три крупных гидрофобных кластера (268, 244 и 60 атомов, как указано выше), которые визуально делят белок на три домена. Один из доменов связан с ДНК, другие два находятся обособленно от первого и менее обособленно друг от друга. Эти два домена участвуют в формировании биологической единицы - димера (см. рисунки далее). Хотелось еще обнаружить эти же домены с помощью алгоритма DETECTIVE, который ищет домены используя предпосылку о том, что у каждого домена есть гидрофобное ядро (ищет гидрофобные ядра по алг. Свинделлса), а потом сопоставить результат с результами CluD, который использует для поиска гидрофобных ядер алгоритм (k,l)-разрезов (k=l=1), но, к сожалению, серверов не нашла используя поисковик google. Что, как не страшно, ведь эти три домена прекрасно видно и их биологическое значение вполне угадывается. Один из доменов участвует в связывании ДНК, а два других помогают формированию димера и, по видимому, являются регуляторными.
Задание 2. Найти гидрофобные кластеры на интерфейсе двух цепочек.
Структура 1VPW имеет только одну цепь А, поэтому возьмем ее биологическую единицу (димер). Чтобы получить димер потребовались команды:
load 1VPW-2.pdb, A1 create A1, A2 alter_state 1,A2,(x,y,z)=(x,-y,-z) save 1vpw_dimer.pdb
| Параметры кластера | Найденные кластеры (остатки) | Иллюстрация | Пояснение к результату |
| 4.5 А, минимальное число атомов 10 | 3 кластера:238,94,10 |  |
Слишком "мягкие" параметры - выделяется почти все |
| 3.5 А, минимальное число атомов 10 | 3 кластера:17,16,15 |  |
найдены гидрофобные кластеры между цепями |
| 4 А, минимальное число атомов 3 | нет | 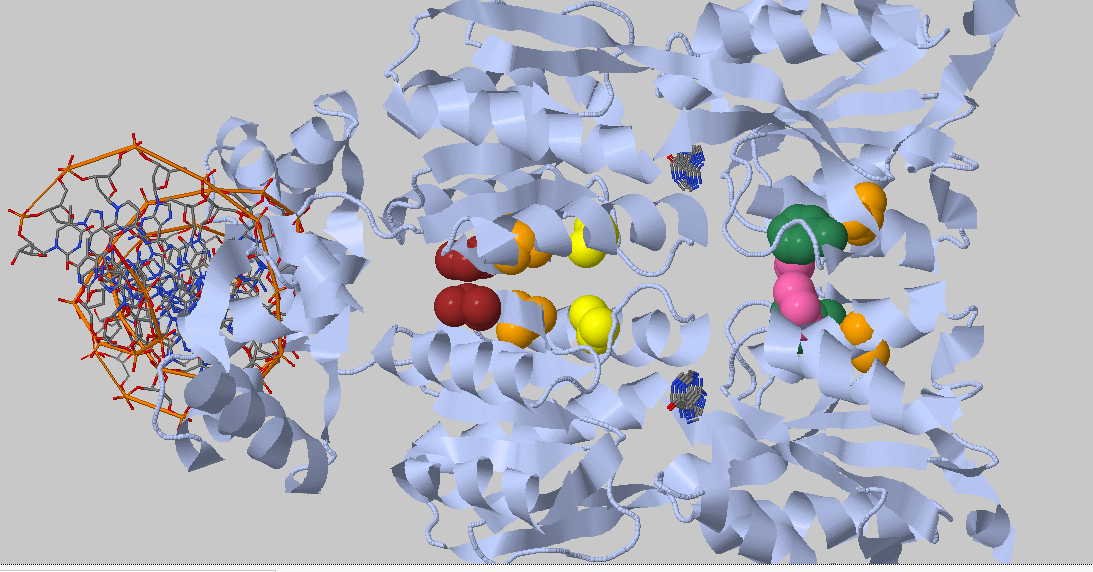 |
слишком "жесткие" параметры, гидрофобные кластеры поделились на кусочки, но уже нет кластеров на интерфейсе двух цепочек (розовые тоже не соединяются) |
Построение поверхности, раскраска участка поверхности: PyMol
Задание 1. Создать изображения для комплекса димера пуринового репрессора с ДНК. Структура 1VPW.
Поверхность контакта мономера белка с симметричным мономером на фоне остовной (ribbon) модели мономера (рис. 14).
 |
| Рис. 14. Поверхность контакта мономера белка с симметричным мономером на фоне остовной (ribbon) модели мономера. |
Поверхность контакта димера белков с двойной спиралью ДНК на фоне остовной модели части белка, вовлечённой в контакт (рис. 15).
 |
| Рис. 15. Поверхность контакта димера белков с двойной спиралью ДНК на фоне остовной модели части белка, вовлечённой в контакт. |
Поверхность контакта ДНК с димером белков на фоне проволочной (sticks) модели двойной спирали (рис. 16).
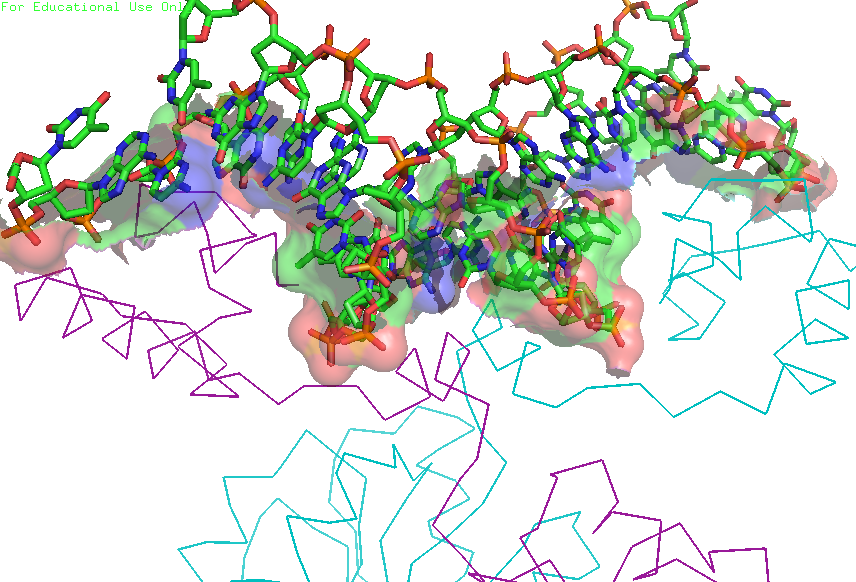 |
| Рис. 16. Поверхность контакта ДНК с димером белков на фоне проволочной (sticks) модели двойной спирали. |
Задание 2. Пользуясь сервисом CluD, определите гидрофобные кластеры объёмом не менее 10 атомов на интерфейсе мономеров белка в том же комплексе.
Возьмем из предыдущего задания результат работы CluD с параметрами 3.5 А, минимальное число атомов 10 (рис. 2 в таблице 2). И выделяем поверхности в PyMol (select surface_hphobic, *_surf and core*) (рис. ).
На сервере CluD можно найти список найденных гидрофобных кластеров и список атомов входящих в них. Чтобы выделить найденные гидрофобные кластеры в PyMol используем команды:
sel core1, c. A and (////103/CB or ////103/CD or ////86/CED2 or ////86/CED1 or ////86/CD2 or ////86/CD1 or ////86/CZ or ////86/CG or ////86/CB or ////107/CZ or ////107/CE1 or ////107/CD2 or ////107/CD1 or ////107/CE2 or ////107/CG or ////107/CB or ////103/CG)
sel core2, c. A and (////252/CB or ////283/CE2 or ////287/CD2 or ////287/CG or ////287/CD1 or ////287/CB or ////283/CD2 or ////283/CE1 or ////283/CD1 or ////283/CG or ////283/CZ or ////283/CB or ////252/CE or ////252/CG or ////252/SD)
sel core3, c. A and (////282/CB or ////282/CD1 or ////282/CE2 or ////282/CD2 or ////223/CB or ////282/CG or ////282/CE1 or ////282/CZ or ////227/CD2 or ////227/CZ or ////227/CE2 or ////257/CB or ////227/CG or ////227/CE1 or ////227/CD1 or ////227/CB)
 |
| Рис. 17. Поверхность контакта мономера белка с симметричным мономером на фоне остовной (ribbon) модели мономера, гидрофобная поверхность выделена красным. |
Сравнение доменов SCOP/SCOPe, ECOD, CATH и Pfam
Задание. Для какого-нибудь белка с известной структурой найдити границы доменов согласно различным классификациям.
Посмотрим данные для белка 3TW2, который встречался и раннее.
SCOPe
Нашел два домена: d3tw2a (3tw2 A:) (A:11-126) и d3tw2b (3tw2 B:) (B:15-126). Классификация по SCOPe:
- Класс: Alpha+beta белки
- Укладка: HIT-like (гистидиновой триады)
- Суперсемейство: HIT
- Семейство: HIT (гистидиновой триады) family of protein kinase-interacting proteins
- Из: Human (Homo sapiens)
CATH
Выделяет тоже два домена: 3tw2A00 (A:11-126) и 3tw2B00 (B:15-126). Классификация по CATH:
- Класс: Alpha beta белки
- Архитектура: двуслойный сэндвич
- Топология: HIT семейство (гистидиновой триады), субъединица А
- Гомология: HIT семейство (гистидиновой триады), субъединица А
ECOD
Выделяет тоже два домена: e3tw2A1 (A:11-126) и e3tw2B1 (B:15-126). Классификация по ECOD:
- A: a+b трехслойные
- X: HIT-like
- H: HIT-related
- T: HIT-related
- F: HIT (семейство гистидиновой триады)
Pfam
Выделяет один домен для обеих цепей: HIT (24-121).
Информация о домене:
- Количество архитектур: 48
- Количество последовательностей: 4038
- Количество видов: 2069
- Количество структур: 131
Таким образом, все базы данных, кроме Pfam, находят одинаковые домены, хотя и называют их и классифицируют их по-своему. Также можно заметить, что эти домены автоматизированно классифицированы, следовательно не очень удивительно, что они совпадают. Так как структура 3TW2 является гомодимером, выделение двух разных доменов, причем разных и по длине представляется бесмысленным. Так эти базы данных могут все подряд делать отдельным доменом, что не есть хорошо. Pfam же нашла один домен для A и В. И этот домен короче, чем найденные в других БД. Поэтому этот домен более общий (домен HIT белков).
Использование сайта PDB
Задание 1. Нужно скачать все последовательности белков, структуры которых определенны при помощи метода электронной микроскопии
Выбираем метод ELECTRON MICROSCOPY с помощью Advanced search. Нашлось 917 структур. Файл с последовательностями.
Задание 2. Необходимо также найдити пару белков, для которых гибкое структурное выравнивание включает существенно больше остатков, чем жесткое.
Воспользуемся сервером FATCAT и выровним структуру 1aj3 (98 из аминокислот) и 2spc (цепь А из 107 аминокислот), пример взят из статьи 2003 года Ye_2003(FATCAT).pdf. Сервер предоставляет возможность выровнить как гибко (рис. 19), так и жестко (рис. 18). Как видно из изображений и по сравнению RMSD, в данном случае гибкое выравнивание существенно лучше: включает больше остатков, чем жесткое (94 vs 67) и имеет ниже значение RMSD (2.02 vs 4.6).
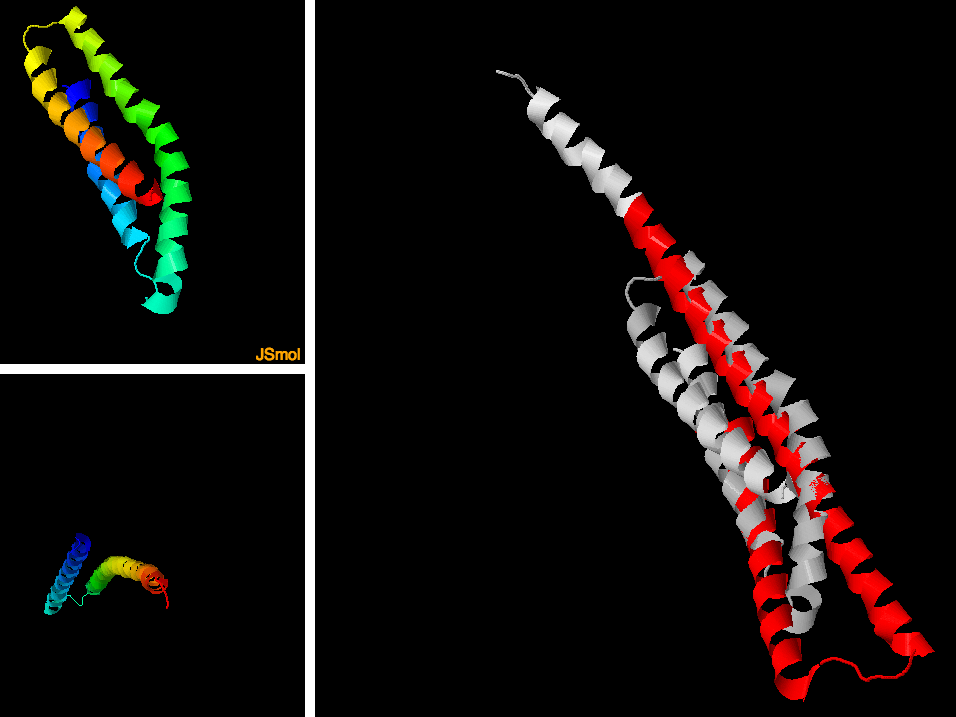 |
| Рис. 18. Жесткое выравнивание 1aj3 и 2spc. Выровнено 67 остатков, RMSD=4.6 (так себе значение), p-value=1.38e-06. |
 |
| Рис. 19. Гибкое выравнивание 1aj3 и 2spc. Выровнено 94 остатка, RMSD=2.02, p-value=1.93e-06, "структуры сильно схожи" (сервер). При выравнивании произошло 2 twist-поворота, один из участков повернулся практически на 180 градусов. |