Навигация по сайту:
|
Анализ транскриптома
Анализ и очистка чтений
Использовавшиеся команды
| Команда |
Комментарий |
| fastqc chr9.1.fastq |
Создает html страницу с отчетом и zip архив с содержимым страницы, а также логи. |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr9.1.fastq trimmed.fastq TRAILING:20 MINLEN:50 |
Убирает из рида нуклеотиды с phred score меньше 20 и удаляет риды, длина которых меньше 50. |
Будем работать с первой репликой (chr9.1.fastq), в ней 19976 ридов. Fastqc жалуется вообще на все, на что только может, но мы ничего не можем с этим сделать.

С другой стороны непонятно, стоит ли на это обращать внимание. Возможно это типичная картина для RNAseq. Совсем короткие и плохие риды отсеялись с помощью trimmomatic, но кардинальных изменений не наступило, разве что в
sequence length distribution.
Результат работы trimmomatic: 19267 (96,45%) Dropped: 709 (3,55%)
Отчет fasqc для исходного файла
Отчет fasqc для обработанного trimmomatic-ом файла
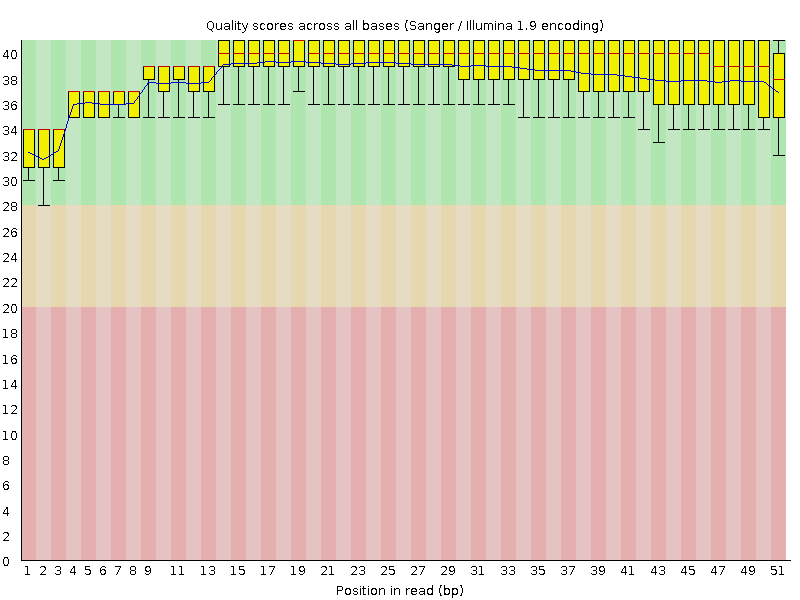 Per base sequence quality до trimmomatic.
Per base sequence quality до trimmomatic.
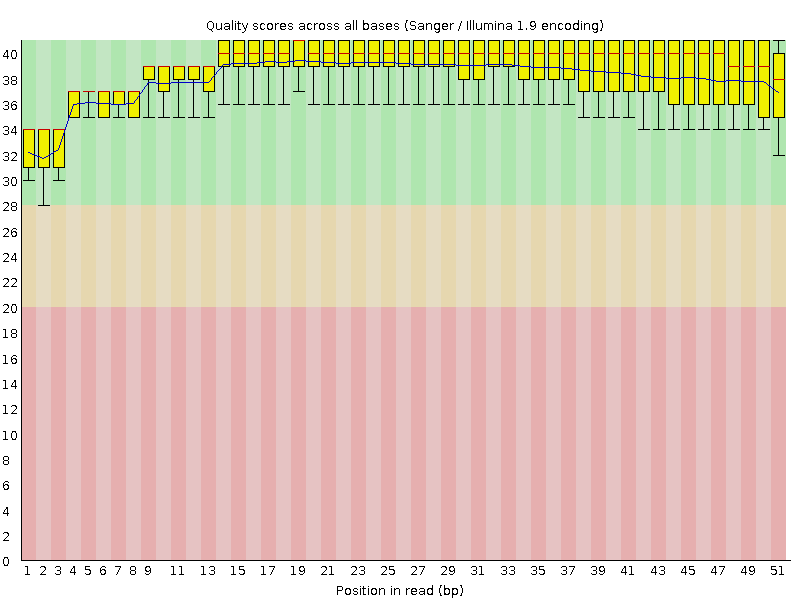 Per base sequence quality после trimmomatic.
Per base sequence quality после trimmomatic.
Картирование ридов
Использовавшиеся команды
| Команда |
Комментарий |
| hisat2 -x ../indexed_genome/chr9 -U trimmed.fastq -S mapped_reads.sam --no-softclip 2> hisat2.log |
Накладывает риды на геном. -х — префикс .ht2 файлов, созданных hisat2-build, -U — файл со чтениями, -S выходной файл .sam,
--no-spliced-alignment не используется, так как секвенировали транскриптом --no-softclip не позволяет наложиться какой-то части рида, игнорируя то, что на концах
,"2>" перенаправляет вывод программы в лог-файл(hisat2 печатает лог на stderr). |
Из прошлого практикума мы уже имеем проиндексированную последовательность девятой хромосомы. --no-splice-alignment отключаем, так как то, что мы секвенировали, в геноме может
оказаться двумя экзонами, разделенными интроном.
Анализ выравнивания
Использовавшиеся команды
| Команда |
Комментарий |
| samtools view -b -o mapped_reads.bam mapped_reads.sam |
Перевод .sam в бинарный вид .bam. |
| samtools sort mapped_reads.bam mapped_reads_sorted |
Сортирует выравнивания ридов в соответствии с их порядком в геноме |
| samtools index mapped_reads_sorted.bam |
Индексация выравниваний |
Вывод hisat2:
19267 reads; of these:
19267 (100.00%) were unpaired; of these:
113 (0.59%) aligned 0 times
19154 (99.41%) aligned exactly 1 time
0 (0.00%) aligned >1 times
99.41% overall alignment rate
Как видно отсюда, большинство(19154) ридов легли на геном ровно один раз. Не были наложены 113 ридов. Более одного раза не
наложился ни один.
Подсчет чтений
Программа htseq-count cчитает сколько ридов упало на данный ген. Некоторые ее опции:
-i GFF атрибут, используемый в качестве feature ID. У нас это gene_id.
-f определяет формат: bam или sam.
-s [yes, no, reverse] сообщает htseq-count относятся ли данные секвенирования исключительно к прямой или исключительно к обратной
цепи. "no" подразумевает, что к обеим.
-m [union, intersection-strict, intersection-nonempty] определяет то, как программа определяет какому гену принадлежат
те или иные риды(бывают спорные ситуации). Смысл union, intersection-strict, intersection-nonempty изложен в картинке ниже.
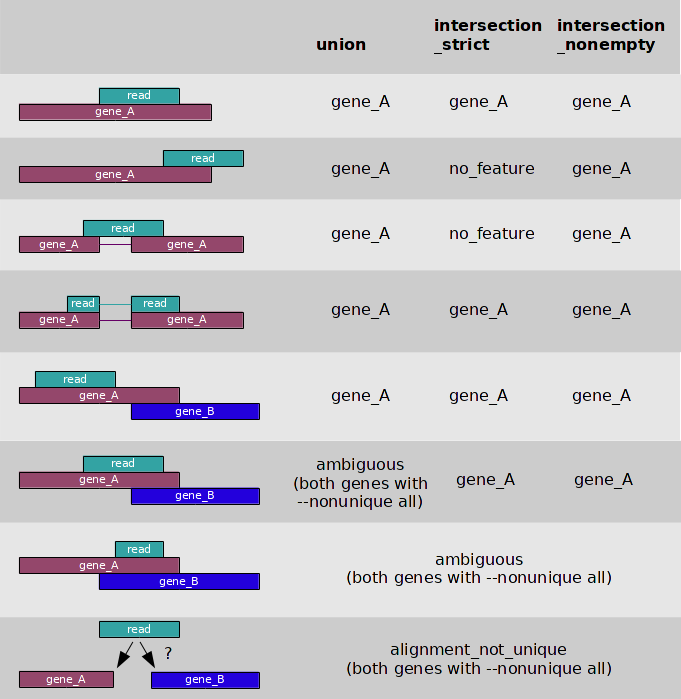 Использовавшиеся команды
Использовавшиеся команды
| Команда |
Комментарий |
| htseq-count -f bam mapped_reads.bam -i gene_id -s no /P/y14/term3/block4/SNP/rnaseq_reads/gencode.v19.chr_patch_hapl_scaff.annotation.gtf > Counts |
Подсчитывает число ридов, упавших на определенный ген. |
| grep -wv 0 Counts > Compact |
Выводит все строки, не кончающиеся на 0 в файл compact. -w переключает в режим совпадений по словам, -v позволяет вывести все, кроме
строк, которые соответствуют регулярному выражению. |
Содержимое файла Compact:
ENSG00000119335.12 18877
__no_feature 277
__not_aligned 113
Большинство ридов принадлежат гену ENSG00000119335.12.
ID ENSG00000119335.12 соответствует ген, кодирующий белок SET nuclear proto-oncogene. Известен тем, что ингибирует ацетилирование нуклеосом и в особенности гистона Н4. Проявляет
анти-апоптотическую активность. Ссылка на UniProt.
Маленькое дополнительное задание: htseq-count с другими параметрами
Результаы intersection-nonempty не отличается от union, intersection-strict действительно strict:
ридов, принадлежащих ENSG00000119335.12 получилось меньше, соотвественно увеличилось __no_feature.
Union:
ENSG00000119335.12 18877
__no_feature 277
__not_aligned 113
intersection-strict:
ENSG00000119335.12 18806
__no_feature 348
__not_aligned 113
intersection-nonempty:
ENSG00000119335.12 18877
__no_feature 277
__not_aligned 113
union, intersection-strict, intersection-nonempty
|