Работа с BLAST
Что было сделано:
- были найдены гомологи моего белка (префлагеллин синтетазы) в базе данных SwissProt
- были объяснены карты сходства двух белков
- были проведены "эксперименты" с BLAST
Задание 1.
Описание параметров BLAST
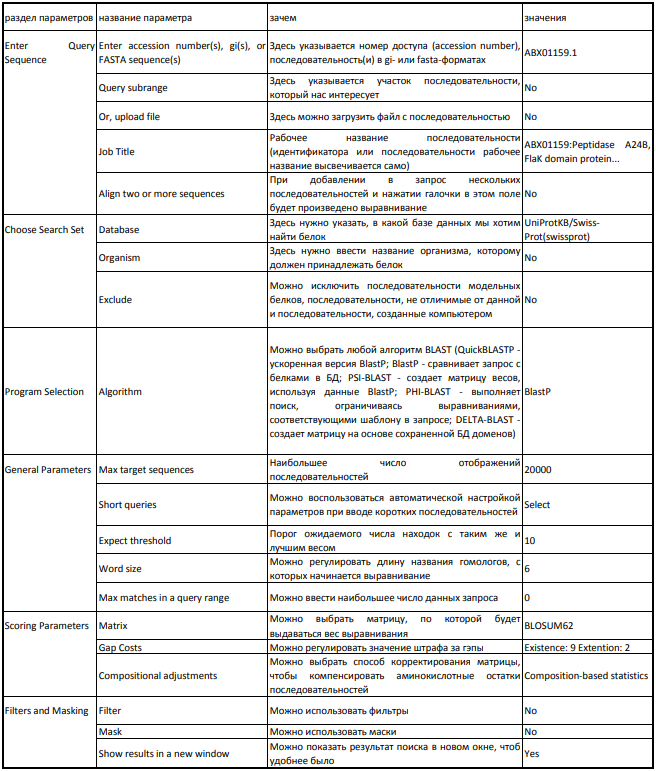
В
результате моего запроса получился банк из шести последовательностей. Подробнее можно посмотреть
в файле скачать файл прямо сейчас
Из них я выбрала четыре, по моему мнению, гомологичных последовательности: A9A677.1, Q6LZR9.1, Q8NKW5.1,
Q58312.2. Я опиралась на название
(род археи Methanococcus), на процент покрытия (у четырех из шести последовательностей этот
показатель оказался очень хорош - не меньше 99%), на процент идентичности (но на него не стоило опираться
в первую очередь, так как этот процент зависел еще и от покрывания: например, последовательность,
составляющаяя 10% от длины запроса, имела практически такой же процент идентичности, что и последовательность,
составляющая 99% от длины запроса) и на Е-value (чем он меньше, тем значимее находка). Также, в качестве
сравнения, я выбрала последовательность, по моему мнению, не гомологичную с запросом: Q5UZH5.2. Она
показалась мне слишком короткой (14% от длины запроса), с самым низким процентом идентичности (30,30%),
самым большим E-value (9,7), да и по названию организма не было совпадений.
Подтверждение вы найдете на странице, пройдя по этой ссылке.
Чтобы проверить мою гипотезу о гомологичности выбранных последовательностей, необходимо было произвести
выравнивание последовательностей, исключая негомологичные (не подходящие по критериям), пока не останутся
одни только гомологичные последовательности. Критерии отбора были такими: во-первых, гомологичные последовательности
что в начале, что в конце должны быть абсолютно идентичными более, чем в 6 колонках подряд; во-вторых,
в гомологичных последовательностях не место колонкам с гэпами; в-третьих, нужно, чтобы в последовательностях
была высокая концентрация идентичных колонок.
В моем случае, гомологичными (т.е. полностью удовлетворяющими критериям) оказались
только две последовательности, из которых одна полностью идентична запросу, только почему-то с другим
номером доступа, а другая оказалась последовательностью аминокислот префлагеллин синтетазы археи
другого штамма.
Здесь
находится множественное выравнивание гомологичных последовательностей.
Задание 2.
Было произведено выравнивание двух последовательностей белков. Первый белок - тетрагидробиопротерин биосинтезирующий ферментоподобный белок организма Gloeophyllum trabeum (трутовый гриб глеофиллум бревенчатый), в таблице обозначен красным цветом и имеет код S7RMD1_GLOTA. Длина последовательности 255 аминокислотных остатков. Второй белок - 7,8-дигидрометилптерин-пирофосфокиназа организма Acanthamoeba castellanii str. Neff (почвенная акантамеба, вызывающая первичный амебный менингоэнцефалит и пневманию), в таблице обозначена зеленым цветом и имеет код L8H299_ACACA. Длина последовательности 636 аминокислотных остатков. Ниже представлена карта выравнивания этих последовательностей.
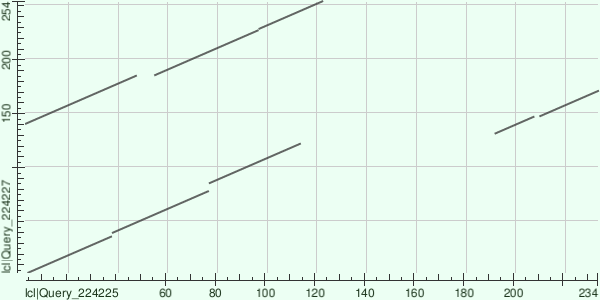
Что мы видим? Для начала, ось последовательности L8H299_ACACA обозначена как Query_224225. Ось последовательности S7RMD1_GLOTA обозначена как Query_224227. В последовательности белка глоефиллума (вертикальная ось) наблюдается дупликация участка последовательности белка акантамебы с 5 до 114 позиции. Также есть инсерция последовательностей на месте разрыва "со смещением" и транслокация последовательности белка акантамебы. Есть небольшие делеции в обеих последовательностях (у белка акантамебы их три, у белка глеофиллума две).
Задание 3.
Давайте поиграем с BLAST. Я набрала выдуманную последовательность "SPRING IS IN THE AIR WHY THEY ASK ME WHERE" и искала последовательности своего белка в Swiss-Prot. Пришлось сначала долго подбирать параметры для того, чтобы в результате появилась хотя бы одна последовательность. Спустя несколько неудачных попыток наконец удалось получить целых две (!) последовательности: Adenine deaminase 2 (АС Q6ANH1.1) и Bile acid-coenzyme A ligase [[Clostridium] scindens] (АС P19409.1). У последовательностей оказались примерно одинаковые веса (27,9 и 25,4 соответственно) и большой E-value (1,1 и 8,4 соответственно). Все подробности можно посмотреть здесь. Вот как я получила такой результат: помимо набора случайной последовательности в поле Enter accession number(s), gi(s), or FASTA sequence(s), я выбрала БД Swiss-Prot (смотри выше), сманила максимальное число отображаемых последовательностей до 20000 (хотя можно было и без этого добиться точно такого же результата), сменила Gap costs на Existense:9 Extention: 2, Composional adjustments поставила на Composition-based statistics, word size - на 3. Все остальные параметры были установлены по умолчанию.
Во второй раз я решила оставить все как и в прошлой сессии поиска (см.выше), но поиск проводила в БД модельных объектов landmark, поменяла параметр Max target sequences на 100. В итоге нашлась одна последовательность с E-value 1,2 и весом 27,9. Имя этой последовательности beta-glucosidase 46 [Glycine max]. Подробнее тут
В третий раз мне захотелось поменять матрицу весов на BLOSUM90. Никакие другие параметры я не меняла. Получился список из восьми модельных последовательностей. Также встретилась последовательность, упомянутая пунктом выше. У нее более, чем в полтора раза уменьшился E-value. Вдобавок, общий вес увеличился на три единицы. Вообще, две цифры в названии матрицы весов означают порог кластеризации. Это значит, например, что в BLOSUM62 все последовательности с идентичностью более 62% объединяются в один кластер, а в BLOSUM90 этот порог выше. Получается, что и результат будет точнее. Это объясняет, почему при замене матрицы весов в результатах поиска появилось гораздо больше последовательностей. С бОльшим количеством последовательностей уменьшается E-value, т.к. последовательность среди увеличенного количества последовательностей становится более редкой. Стоит отметить, что среди последовательностей, предоставленных BLAST в данном сеансе, есть целых четрые последовательности с одинаковым процентом идентичности и покрывания. Это последовательности с гомологичными белками - факторами элонгации G. Подробнее о результатах поиска тут.

