Ресеквенирование. Поиск полиморфизмов у человека
Что было сделано:
- была проиндексирована референсная последовательность четвертой хромосомы
- было проконтролировано качество чтений с помощью программы FastQC
- была произведена очистка чтений с помощью программы Trimmomatic
- были картированы чтения после процедуры триммирования
- было поизведено картирование триммированных чтений
- был произведен анализ выравнивания триммированных чтений и референса
- был проведен поиск snp и инделей
Номер хромосомы: 4
| Использованная команда | Описание |
|---|---|
| hisat2-build chr4.fasta index | строит индекс HISAT2 из набора последовательностей ДНК *HISAT2 - это быстрая и чувствительная программа выравнивания для картирования считываний секвенирования нового поколения (данных о целом геноме, транскриптоме и экзоме) по общей популяции людей (а также по одному эталонному геному). |
| fastqc chr4.fastq | удаленный запуск программы FastQC, проводящей анализ качества чтений выходной файл в формате архива, содержащего html-страницу с результатом |
| java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr4.fastq 4out.fastq TRAILING:20 MINLEN:50 | запуск программы Trimmomatic, проводящей очистку чтений, на kodomo TRAILING:20 отрезает базы от конца чтения, если качество ниже порогового, шаг обрезки в программе Trimmomatic MINLEN:50 сбрасывает чтение, если оно меньше указанной длины, шаг обрезки в программе Trimmomatic |
| fastqc 4out.fastq | удаленный запуск программы FastQC, проводящей анализ качества чтений выходной файл в формате архива, содержащего html-страницу с результатом |
| hisat2-build chr4.fasta chr4 | Индексация референсной последовательности |
| hisat2 -x chr4 -U 4out.fastq -S 1sam.sam --no-spliced-alignment --no-softclip | выравнивание прочтений и референса в формате .sam, запуск hisat2 с параметрами --no-spliced-alignment и --no-softclip и сохранение результатов в отдельном файле |
| samtools view 1sam.sam -b >> 1bam.bam | Перевод выравнивания чтений с референсом в бинарный формат .bam с помощью команды view пакета samtools |
| samtools sort 1bam.bam 2bam | Сортировка выравнивания чтений с референсом по координате в референсе начала чтения |
| samtools index 2bam.bam | Индексирование отсортированного .bam файла |
| samtools mpileup -uf chr4.fasta -g -o snp.bcf 2bam.bam | создание файла с полиморфизмами в формате .bcf |
| bcftools call -cv -o snp2.vcf snp.bcf | создание файла со списком отличий между референсом и чтениями в формате .vcf |
| convert2annovar.pl -format vcf4 2snp.vcf > chr4.avinput | Конвертирование файла со списком отличий референса и чтений для программы annovar |
| annotate_variation.pl | Запуск скрипта для аннотации файла с snp с помощью предложенных баз данных |
| annotate_variation.pl -filter -out dbsnp -build hg19 -dbtype snp138 chr4.avinput /nfs/srv/databases/annovar/humandb.old | Аннотация по Dbsnp |
| annotate_variation.pl -out chr4_refgene -build hg19 chr4.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотация по Refgene |
| annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out 1000g chr4.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотация по 1000 Genomes |
| annotate_variation.pl -regionanno -build hg19 -out gwas -dbtype gwasCatalog chr4.avinput /nfs/srv/databases/annovar/humandb.old/ | Аннотация по GWAS |
| annotate_variation.pl chr4.avinput -filter -dbtype clinvar_20140211 -buildver hg19 -out clinvar /nfs/srv/databases/annovar/humandb.old/ | Аннотация по ClinVar |
Количество исходных чтений: 5810
Результаты работы с FastQC:
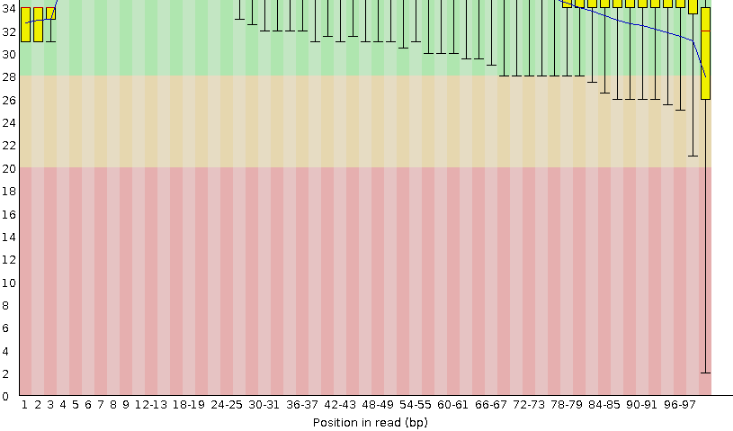
ссылка на страницу с результатами работы программы
Осталось чтений после процедуры триммирования: 5715. В случае моих чтений процедура триммирования была оправдана. Качество ридов повысилось, подтверждение можно посмотреть на картинке ниже.
Качество чтений FastQC после триммирования:
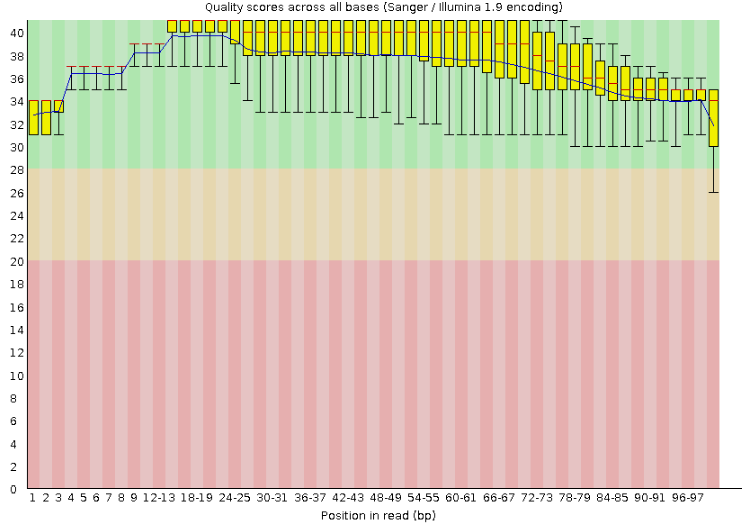
ссылка на страницу с результатами работы программы
Процент чтений, картированных на геном: 99.67%
Вывод программы Hisat2:
5715 reads; of these: 5715 (100.00%) were unpaired; of these: 19 (0.33%) aligned 0 times 5695 (99.65%) aligned exactly 1 time 1 (0.02%) aligned >1 times 99.67% overall alignment rate
Качество картирования: Судя по результатам программы Hisat2, качество картирования достаточно хорошее.
Описание трех полиморфизмов из .vcf файла:
Полиморфизм №1
- Координата: 68426172
- Тип полиморфизма: замена
- Референс - чтение: А - С
- Глубина покрытия места: 1
- Качество чтений в данном месте: 4.77219
Полиморфизм №2
- Координата: 68458937
- Тип полиморфизма: замена
- Референс - чтение: G - C
- Глубина покрытия места: 87
- Качество чтений в данном месте: 225.009
Полиморфизм №3
- Координата: 88757565
- Тип полиморфизма: вставка
- Референс - чтение: ATGTGTGTGTGTGTGTGTGT - ATGTGTGTGTGTGTGTGTGTGT
- Глубина покрытия места: 1
- Качество чтений в данном месте: 17.548
Количество snp: 45
Количество инделей: 4
Оценка покрытия и качества полиморфизмов:
Категории snp в annovar базы данных Refgene: intronic, intergenic, exonic, downstream, UTR3
Количество snp, попавших в каждую категорию:
- intronic - 39
- intergenic - 3
- exonic - 3
- downstream - 2
- UTR3 - 1
Гены, в которые попали snp:
- KLKB1 - 18 snp (kallikrein B1)
- MEPE - 16 snp (matrix extracellular phosphoglycoprotein)
- STAP1 - 12 snp (signal transducing adaptor family member 1)
- KIAA0922 - 3 snp (Transmembrane protein 131-like)
Сколько snp имеет rs: 43
Частота найденных snp: максимальная = 0.91853, минимальная - 0.00379393
Клиническая аннотация snp:
Вот что выдало аннотирование по базе данных GWAS:
gwasCatalog Name=Parkinson's disease chr4 68447249 68447249 A G het 176.009 24 gwasCatalog Name=Cardiovascular disease risk factors chr4 88755828 88755828 T C hom 221.999 99 gwasCatalog Name=Metabolite levels chr4 187149540 187149540 G A hom 212.999 15 gwasCatalog Name=Obesity-related traits chr4 187158034 187158034 G A hom 221.999 83
Имеется четыре полиморфизма, представленные заменой одного нуклеотида, ведущие к следующим заболеваниям:
- болезнь Паркинсона
- фактор риска кардиоваскулярной болезни
- уровень метаболитов
- черты, связанные с ожирением
Вот этот результат выявило аннотирование по БД ClinVar:
CLINSIG=pathogenic;CLNDBN=Prekallikrein_deficiency;CLNREVSTAT=no_assertion_criteria_provided;CLNACC=RCV000012817.24;CLNDSDB=MedGen:OMIM:SNOMED_CT;CLNDSDBID=C0272339:612423:48976006 chr4 187158034 187158034 G A hom 221.999
Выявлен один патогенный однонуклеотидный полиморфизм, ведущий к дефициту прекалликреина - состоянию крови, обычно не вызывающему серьезных проблем со здоровьем.

