Анализ транскриптомов
Что было сделано:
- был проведен анализ качества чтений
- было произведено картирование чтений
- был проведен анализ выравниваний
- был проведен подсчет чтений
Задание 1.
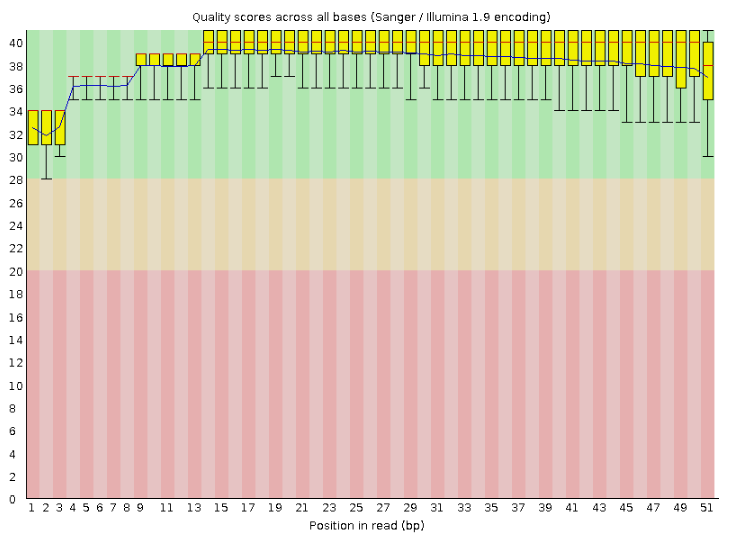
Были даны файлы с чтениями. Взяла первую биологическую реплику. Ниже приведен результат в виде картинки. Ссылка на html-страницу здесь.
| Команда | Описание |
|---|---|
| fastqc chr4.1.fastq | проводит контроль качества чтений |
Задание 2.
При проведении картирования чтений нужно было сделать ту же самую процедуру с hisat2, но убрать при этом один из параметров. Убран парраметр --no-spliced-alignment, так как в данном случае рассматривается зрелая РНК, в которой рядом могут находиться участки, которые изначально не были соседними.
| Команда | Описание |
|---|---|
| hisat2-build chr4.fasta chr4 | Индексация референсной последовательности (можно было не использовать, т.к. использовались те же индексные файлы, что и при картировании экзомного секвенирования ) |
| hisat2 -x chr4 -U chr4.1.fastq -S chr4.1_sam.sam --no-softclip | Выравнивание чтений и референса в формате .sam, запуск hisat2 с параметром --no-softclip и сохранение результатов в отдельном файле |
Выдача команды hisat2:
2735 reads; of these: 2735 (100.00%) were unpaired; of these: 72 (2.63%) aligned 0 times 2663 (97.37%) aligned exactly 1 time 0 (0.00%) aligned >1 times 97.37% overall alignment rate
Задание 3.
| Команда | Описание |
|---|---|
| samtools view chr4.1_sam.sam -b >> chr4.1_bam.bam | Перевод выравнивания чтений с референсом в бинарный формат .bam с помощью команды view пакета samtools |
| samtools sort chr4.1_sam.sam chr4.1_bam2 | Сортировка выравнивания чтений с референсом по координате в референсе начала чтения |
| samtools index chr4.1_bam2.bam | Индексирование отсортированного .bam файла |
| samtools idxstats chr4.1_bam2.bam > chr4.1.txt | Количество откартированных чтений |
Задание 4.
| Команда (опция) | Описание |
|---|---|
| htseq-count | принимает один/более файлов выравниваний в формате .sam или .bam и файл особенностей в формате .gff. В результате для каждой фичи выдаёт количество ридов, откартированных на неё |
| -s {yes,no,reverse}, --stranded {yes,no,reverse} | Проверяет, являются ли данные анализа специфичными для конкретной цепи, (по умолчанию yes), 'reverse' означает 'yes' с обратной интерпретацией цепей |
| -i IDATTR, --idattr IDATTR | GFF атрибут, используется как feature ID (default, suitable for Ensembl GTF files: gene_id) |
| -f {sam,bam}, --format {sam,bam} | Формат принимаемого файла, по умолчанию .bam |
| -m {union,intersection-strict,intersection-nonempty}, --mode {union,intersection-strict,intersection-nonempty} | опция для перекрывающихся чтений, по умолчанию union |
| htseq-count -s no -f bam chr4.1_bam2.bam -m intersection-nonempty -i gene_id /nfs/srv/databases/ngs/spyro/gencode.v19.chr_patch_hapl_scaff.annotation.gtf > chr4.1_htseq-count.out | Kоличество ридов, выровненных с разными участками референса: -s no входные данные необязательно были с одной цепи; -m intersection-nonempty пересечение только не пустых feature |
Задание 5.
| Команда | Описание |
|---|---|
| grep -wv 0 chr4.1_htseq-count.out | Поиск ненулевых строк |
Выдача команды:
ENSG00000071127.12 1884 ENSG00000223086.1 1 ENSG00000261490.1 2 __no_feature 776 __not_aligned 72
1884 рида легли в основу WDR1 (ENSG00000071127.12). Этот ген кодирует белок, содержащий 9 повторов WD. WD-повторы представляют собой приблизительно 30-40-аминокислотные домены, содержащие несколько консервативных остатков, в основном, включая trp-asp на С-конце. Домены WD участвуют в межбелковых взаимодействиях. Кодированный белок может помочь вызвать разборку актиновых филаментов. Посмотреть подробнее об этом гене можно здесь.
776 ридов не легли в границы генов, а 72 не откартировались.

