EMBOSS
Упражнения
Выполнил следующие упражнения:
- Собрал несколько .fasta файлов в один:
sstarikov@kodomo:~/public_html/term3/block2$ ls *.fasta > list
sstarikov@kodomo:~/public_html/term3/block2$ seqret @list sequences.fasta
- Разделил файл с несколькими последовательностями на несколько .fasta файлов:
sstarikov@kodomo:~/public_html/term3/block2$ seqretsplit some.fasta seq.fasta
Read sequences and write them to individual files
- Транслировал последовательность в шести рамках:
sstarikov@kodomo:~/public_html/term3/block2$ transeq final.fasta frames.fasta -frame 6
Translate nucleic acid sequences
- Перевел выравнивание из fasta формата в формат .msf
sstarikov@kodomo:~/public_html/term3/block2$ seqret somealignment.fasta msf::somealignment.msf
Read and write (return) sequences
- Нашел частоты кодонов в данных кодирующих последовательностях:
sstarikov@kodomo:~/public_html/term3/block2$ cusp final.fasta freq.cusp
Create a codon usage table from nucleotide sequence(s)
Получился файл freq.cusp с таблицей, в которой указаны кодоны, какие аминокислоты они кодируют, сколько было найдено кодонов каждого типа и какой процент от общего числа они составляют.
Сравнение аннотации генов белков в хромосоме бактерии Dyadobacter fermentans DSM 18053 с трансляциями длинных открытых рамок считывания
Бактерия - Dyadobacter fermentans DSM 18053 (NC_013037.1, CP001619.1)
Я получил список координат и ориентаций найденных открытых рамок так:
getorf NC_013037.gb orfs.fasta -table 11 -minsize 180 -find 1 -circular
-table таблица генетического кода, здесь 11 (бактериальная таблица генетического кода)
-minsize минимальная длина открытой рамки
-find 1 позволяет транслировать найденные рамки от старт-кодона до стоп-кодона
-circular данная молекула ДНК имеет кольцевую структуру
Я получил файл с трансляциями длинных открытых рамок с помощью программы infoseq так:
infoseq orfs.fasta -outfile info.txt -only -name -length -description
Затем я преобразовал файл в .xlsx и отсортировал по старту: orfs.xlsx
Затем я создал файл с аннотированными генами белков: prot.xlsx
Последовательности белков - в файле protein_seq.fasta
Затем я создал объединенную таблицу и отсортировал ее: fullsorted.xlsx
Я сделал следующие наблюдения:
- Открытых рамок найдено в 7 раз больше, чем аннотированных генов белков.
Это можно объяснить тем, что далеко не все рамки являются генами, а также тем, что в таблице представлены только аннотированные гены белков (нет генов РНК,...).
- ORF, соответствующие генам белков, обычно длиннее генов белков. (см. 5)

- ORF, соответствующие генам белков, обычно заканчиваются на 3 нуклеотида раньше, чем гены белков (координаты ORF - не включают стоп-кодон).


- Не для всех белков были найдены ORF - не найдены для многих длинных генов.
Причина, скорее всего, в том, что я определял ORF от старт до стоп кодонов, таких ORF получилось много, причем коротких. Деталей алгоритма поиска ORF не знаю, поэтому не могу сказать, как именно он находил ORF.
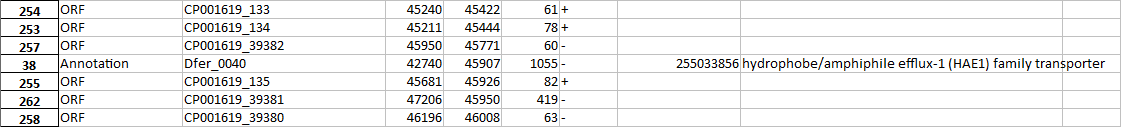
- Открытые рамки считывания начинаются раньше обычно на пару десятков нуклеотидов, чем соответствующие аннотированные гены.


- Из-за порога длины ORF величиной 180 bp не были найдены рамки для коротких аннотированных белков (до 60 aa).


Антипараллельные рамки
Примеры с перекрыванием более 150 п.н. (а таких примеров очень много):
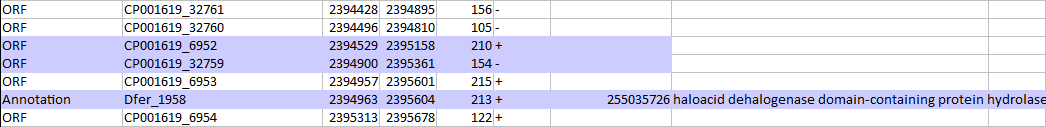
Рамка на обратной цепи перекрывается с геном белка.
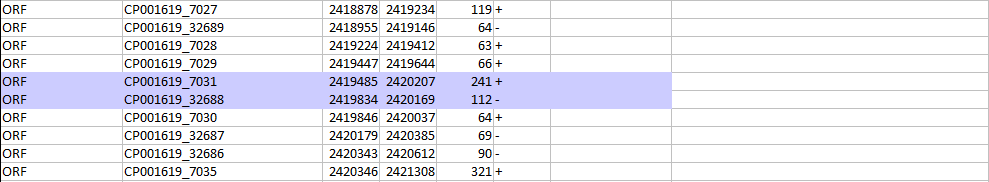
Рамка на обратной цепи не перекрывается с генами белков.
Моя главная страница
© Sergey Starikov, 2015