Я работал с 19 хромосомой и ее ридами
Все использованные команды приведены в конце страницы.
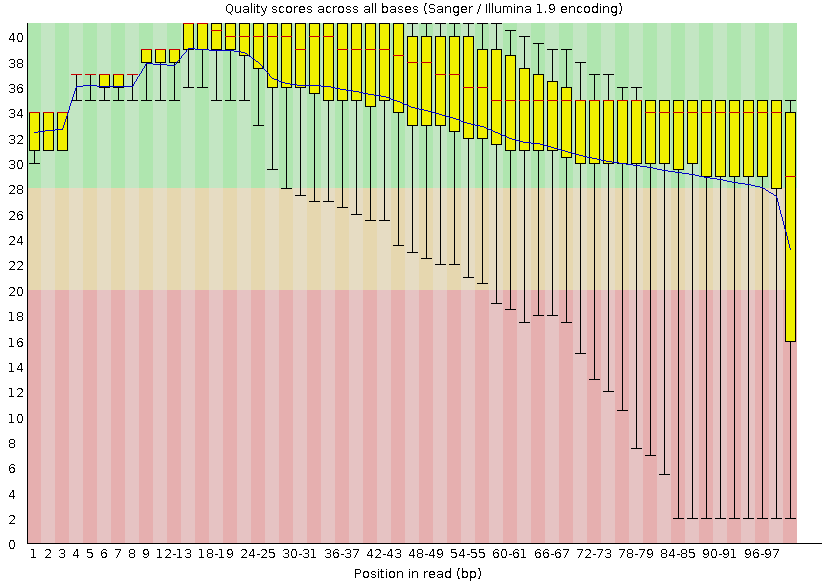
Fastqc before trimming:
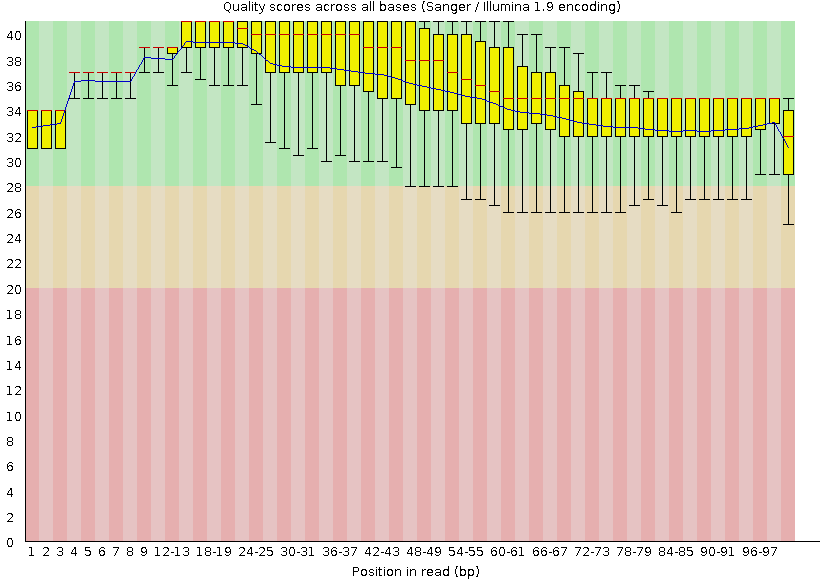
Fastqc after trimming
Число ридов до очистки - 5524, после - 5227 (отброшено 297)
Cначала параметр TRAILING:20 привел к отрезанию нуклеотидов с плохим качеством с конца, затем параметр MINLEN:50 - к удалению ридов несоответствующей длины (меньше 50 п.о.)
Команды приведены в конце страницы.

Откартировались на хромосому все риды.
Найдено 92 полиморфизма, 6 из них - индели, остальные 86 - однонуклеотидные замены.
| Координата | Тип полиморфизма | Референс | Чтения | Глубина покрытия | Качество чтений |
| 17264961 | замена | G | C | 22 | 153.008 |
| 17273753 | замена | C | T | 31 | 148.008 |
| 17283383 | вставка | AGG | AGGG | 9 | 168.492 |
Клиническая аннотация SNP такова - некоторые SNP были в ходе GWAS ассоциированы с ростом, рассеянным склерозом, болезнью Альцгеймера и метаболическим синдромом, но в clinvar не попали, т.е. не была достаточным образом подтверждена их значимость в развитии заболеваний.
Вся полученная информация была сведена в одну Таблицу .xls
Или второй вариант - .xlsx Таблица .xlsx
Или третий вариант - .ods Таблица .ods
Или четвертый вариант (без желтой 64 строки) - .csv Таблица .csv
Выделенный желтым SNP представляет интерес, т.к. экзонный, есть в gwas, замена несинонимичная
| Команда | Зачем |
| fastqc chr19.fastq | анализ качества чтений |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr19.fastq chr19-trimmed.fastq TRAILING:20 MINLEN:50 | очиcтка чтений: удаление нуклеотидов с качеством ниже 20, затем удаление чтений длиной меньше 50 |
| bwa index chr19.fasta | индексирование референсной последовательности |
| bwa mem chr19.fasta chr19-trimmed.fastq > chr19.sam | выравнивание очищенных чтений с проиндексированной референсной последовательностью |
| samtools view chr19.sam -b -o chr19.bam | перевод выравнивания в бинарный формат |
| samtools sort -T /tmp/chr19_sorted -o chr19_sorted.bam chr19.bam | сортировка выравнивания по координате начала чтения в референсной последовательности |
| samtools index chr19_sorted.bam | индексирование отсортированного файла |
| samtools idxstats chr19_sorted.bam | для определения, сколько чтений откартировалось |
| samtools mpileup -uf chr119.fasta chr19_sorted.bam -o chr19snp.bcf | создание файла с полиморфизмами |
| bcftools call -cv chr19snp.bcf -o chr19snp.vcf | перевод .bcf файла в .vcf формат (в список отличий) |
| perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 chr19snp.vcf -outfile chr19snp.avinput | создание файла для annovar из .vcf файла |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -geneanno -dbtype refGene -buildver hg19 chr19snp.avinput -outfile chr19snp_refgene /nfs/srv/databases/annovar/humandb/ | аннотация по базе данных refgene (hg19) |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype snp138 -buildver hg19 chr19snp.avinput -outfile chr19snp_snp138 /nfs/srv/databases/annovar/humandb/ | аннотация по базе данных dbsnp |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 chr19snp.avinput -outfile chr19snp_1000g2014oct /nfs/srv/databases/annovar/humandb/ | аннотация по базе данных 1000genomes |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -dbtype gwasCatalog -buildver hg19 chr19snp.avinput -outfile chr19snp_gwas /nfs/srv/databases/annovar/humandb/ | аннотация по базе данных gwas |
| perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 chr19snp.avinput -outfile chr19snp_clinvar /nfs/srv/databases/annovar/humandb/ | аннотация по базе данных clinvar |