Практикум 12
Сравните выравнивания одних и тех же последовательностей разными программами
Для множественного выравнивания я взяла белки из 9 практикума. Вот они:
- ACCD_ARATH
- ACCD_CUSGR
- ACCD_SOLLC
- ACCD_ADICA
- ACCD_SOLTU
В качестве программ для множественного выравнивания я использовала: Clustal Omega, Kalign, Mafft и T-Coffee.
С помощью программы Jalview
Есть два участка выравнивания, где все 4 программы нвыравнивают примерно одинаково: 240-300 и 410-580. Вероятно это более консервативные участки. Относительно других участков алгоритмы сильно расходятся. Вероятно такие участки менее консерватины и разные алгоритмы будут выравнивать их по разному.
С помощью программы Лизы
Доля одинаково выравненных позиций составила 38% для MAFFT и Kalign и 40% для T-Coffee и Clustal Omega. В выравнивание есть обширные неконсервативные участки — отсюда относительно небольшая доля одинаково выровненных участков.
Ссылки на выдачи
- Выравнивание в Jalview
- Вывод программы Лизы для выравниваний MAFFT и Kalign
- Вывод программы Лизы для выравниваний T-Coffee и Clustal Omega
- Вывод программы VerAlign для выравниваний MAFFT и Kalign
- Вывод программы VerAlign для выравниваний T-Coffee и Clustal Omega
Выравнивание по совмещению структур
Для совмещения структур я взяла семейство белков лептинов. Этот белок является гормоном, участвующим в энергетическом обмене. У человека он подавляет чувство голода.
Для выравнивания я взяла 3 лептина: LEP_PANTR (лептин шимпанзе), LEP_GORGO (лептин гориллы), LEP_PONPY (лептин орангутана).
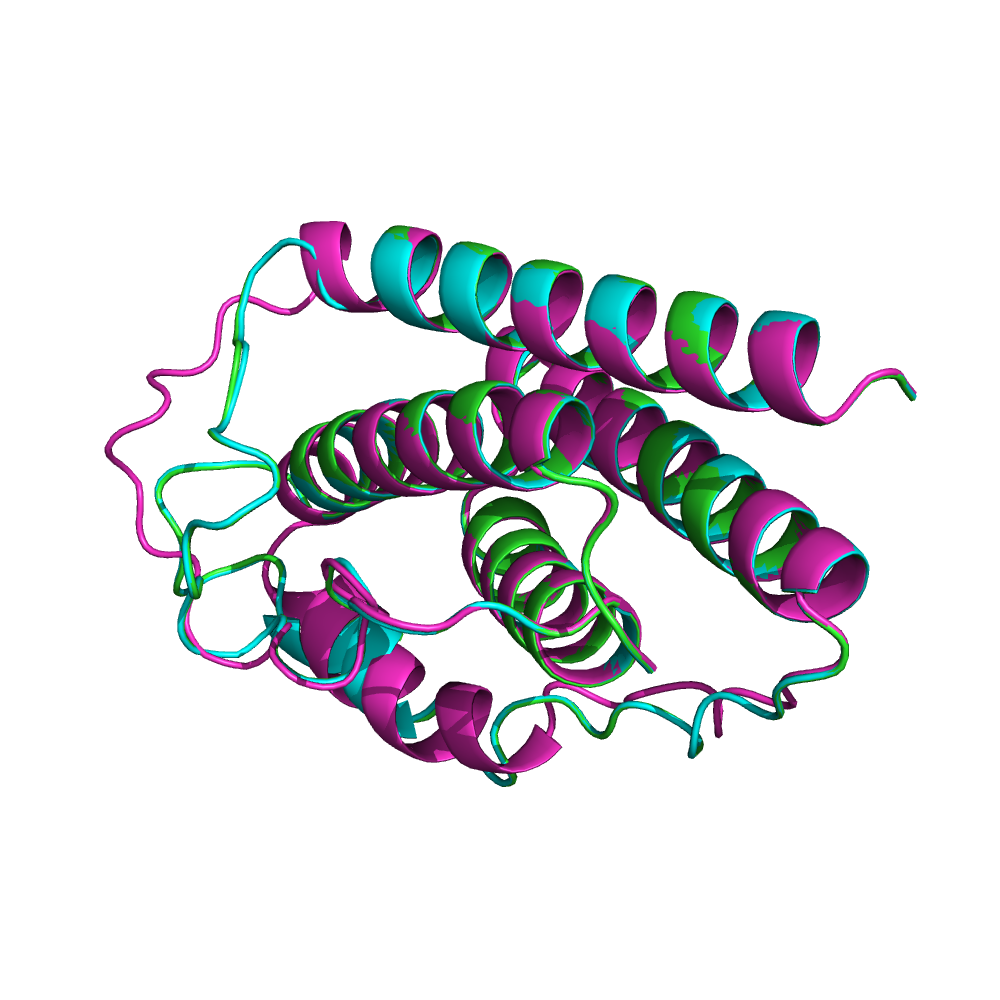
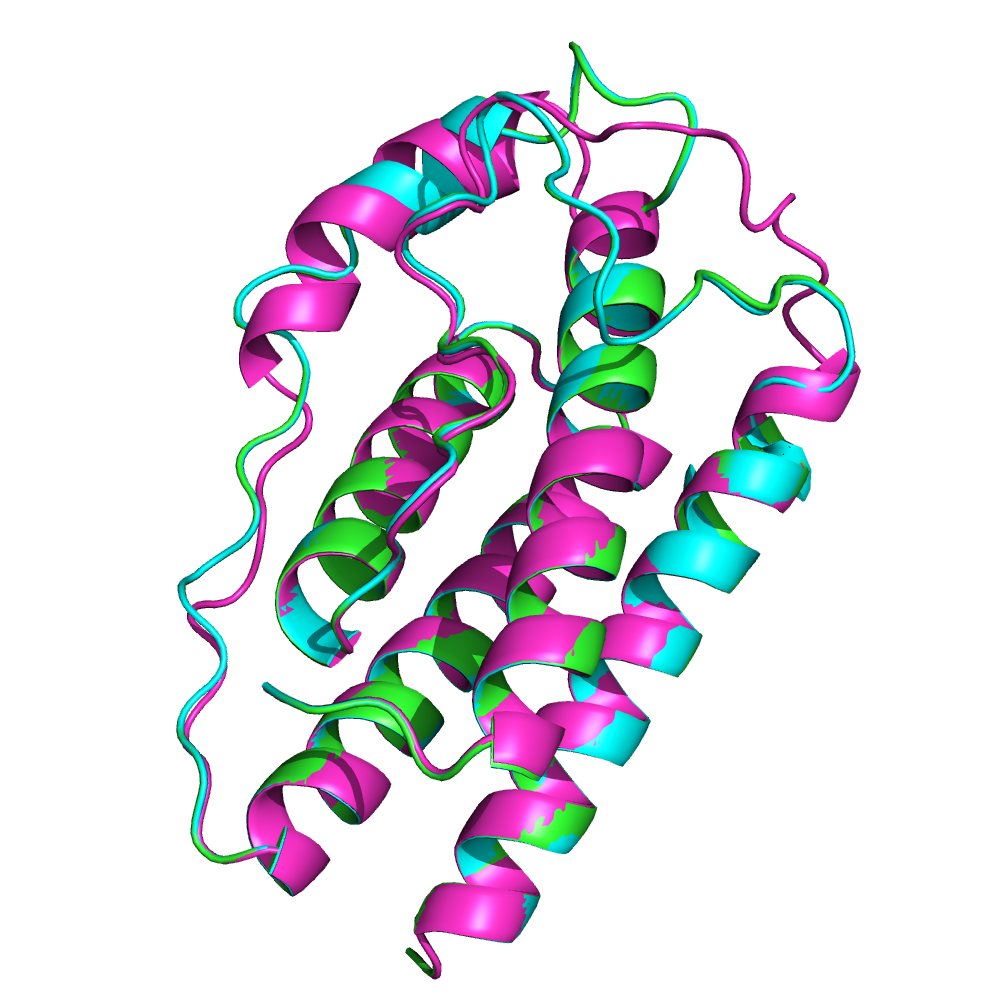
В общем лептины очень похожи. У всех трех сохраняются пять альфа спиралей, как и в целом пространственная структура белка.
Лептины гориллы и шимпазе более схожи между собой в структурном выравнивание. В MSA оказалось что данные лептины отличаются только на 1 аминокислоту, а лептин орангутана отличается от них на 5 аминокислот.
Выравнивание MAFFT в Jalview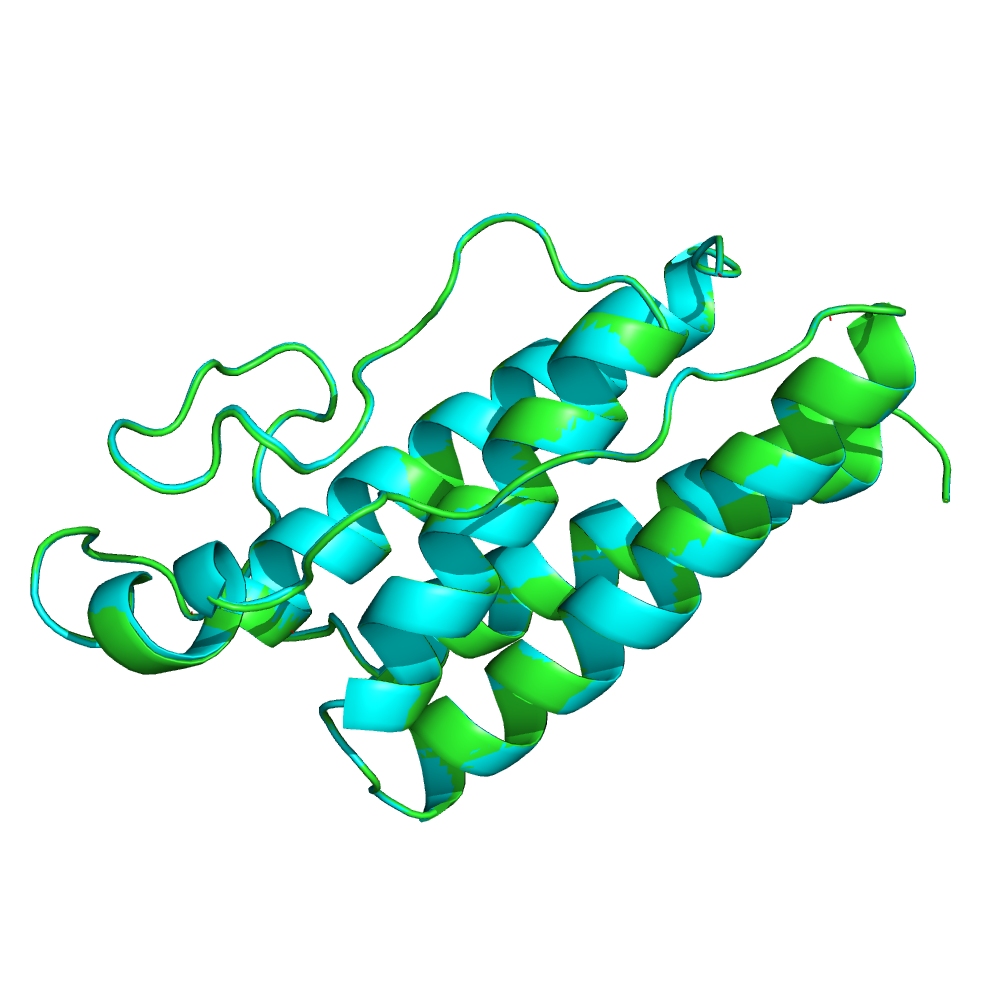

Рассмотрим какие замены произошли в лептине орангутана и как они повлияли на пространственную структуру. В 131 позиции у лептинов шимпанзе и гориллы находится небольшая аминокислота глицин, в то время как у орангутана — аргинин, заметно более большая аминокислота, к тому же заряженная.

Забавная замены произошли в позициях 17-18: валин и изолейцин будто поменялись местами.
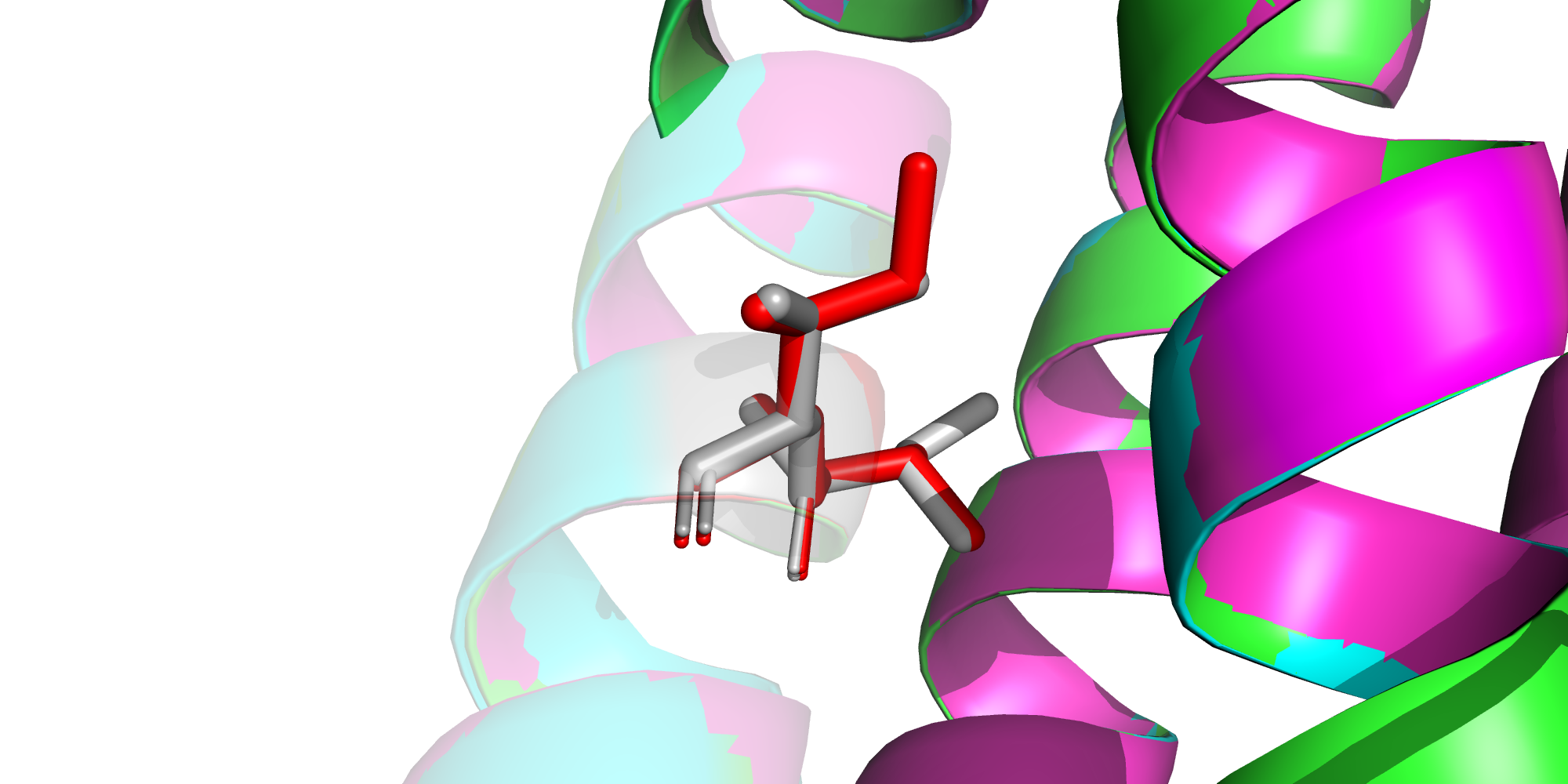
В целом нет ничего необычного в том, что одна гидрофобная аминокислота поменялась на другую. По размеру валин и изолейцин также отличаются не сильно, поэтому значительного изменения пространственной структуры соседних альфа спиралей не наблюдается.
Краткое описание Clustal Omega
Clustal Omega является последней версией широко используемого пакета Clustal MSA. Особенность программы в том, что она позволяет выравнивать досточно большое число последовательностей за небольшое время, при этом точность выравнивания остается высокой. К примеру, на выравнивание более 190 000 последовательностей уйдет всего несколько часов (используя одноядерный процессор). На тестах эта программа показала более точные результаты, чем широко используемые быстрые методы выравнивания.
В предшествующих алгоритмах направляющее дерево строилось путем выравнивания всех N последовательностей между собой попарно. Такой алгоритм имеет сложность О(N^2), из-за чего большое количество последователностей (> 10 000) будут выравниваться крайне медленно.В Clustal Omega используется модифицированный алгоритм построения направляющего дерева, который является O(Nlog(N)^2) сложным алгоритмом. Основывается он на методе эмбеддингов. Последовательности сопаставляются с набором векторов в некотором t-мерном пространстве эмбеддингов. Построение эмбедингов выполняется таким образом, чтобы расположение векторов наилучшим образом соответствовало отношению между исходными последовательностями. Расстояние между получившимися векторами рассчитываются намного быстрее и менее затратно по памяти, чем классическое попарное выравнивание. Дальше попарные расстояния кластеризуются с использованием метода K-средних. И затем в Clustal Omega используется пакет HHalign для выравнивания последовательностей в соответсвие с направляющим деревом от листьев к корню.
Источники
- Sievers, F., & Higgins, D. G. (2013). Clustal Omega, Accurate Alignment of Very Large Numbers of Sequences. Multiple Sequence Alignment Methods, 105–116. doi:10.1007/978-1-62703-646-7_6 (https://doi.org/10.1007/978-1-62703-646-7_6)
- Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., … Higgins, D. G. (2014). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology, 7(1), 539–539. doi:10.1038/msb.2011.75 (https://doi.org/10.1038/msb.2011.75)