Практикум 8
Описание сигнала
Для этого задания я взяла сигнал ядерной локализации, он же NLS. Его узнают транспортные факторы — кариоферины (они же импортины), после чего осуществляется перенос в ядро. Сигнал имеет разные последовательности,
лучше всего исследованы классические сигналы ядерной локализации. Белки с классическими NLS распознаются и связываются кариоферином α при этом сам сигнал может быть одночастным или двухчастным.
Одночастные сигналы состоят из одного участка основных аминокислот, состоящих преимущественно из остатков лизина (K) и аргинина (R). Примером моночастичного NLS является NLS большого Т-антигена вируса SV40: 126 PKKKRKV 132.
Аминокислотная замена второго лизина (выделен жирным) полностью прекращает ядерный импорт, что подчеркивает важность этого остатка. Двухчастные последовательности содержат два кластера основных аминокислот, разделенных линкерной областью неконсервативных аминокислот (10-13 остатков).
Архетипическая двучастная NLS содержится в белке Xenopus laevis — нуклеоплазмине: 155 KRPAATKKAGQAKKK 169.
Ниже я привела консенсусные последовательности классических NLS, а также группу сигналов ядерной локализации, называемую PY-NLS:

Сигнал ядерной локализации должен быть сильным для белков, функционирующих исключительно в кариоплазме.
PWM
Для построения PWM была выбрана последовательность Kozak человека. Для обучения и тестирования я взяла последовательности начала генов X хромосомы с прямой цепи. Для
этого я сначала отфильтровала координаты начала генов из файла с координатами всех генов:
import pandas as pd
all_genes = pd.read_csv("/content/human-genes.tsv", sep="\t")
start_list = all_genes[(all_genes["strand"] == "+") & (all_genes["#chrom"] == "chrX")]["thickStart"].to_list()[:200]
import requests, sys
server = 'https://rest.ensembl.org'
seq = []
for coord in start:
ext = f"/sequence/region/human/1:{coord-6}..{coord+6}:1?expand_3prime=0;expand_5prime=0"
r = requests.get(server+ext, headers={ "Content-Type" : "text/x-fasta"})
if not r.ok:
r.raise_for_status()
sys.exit()
seq.append(r.text.split('\n')[1])
import numpy as np
train = seq[0:100]
test = seq[100:200]
train_spl = np.array([list(i) for i in train])
test_spl = np.array([list(i) for i in test])
mask_A = train_spl == 'A'
count_A = mask_A.sum(axis=0)
mask_T = train_spl == 'T'
count_T = mask_T.sum(axis=0)
mask_G = train_spl == 'G'
count_G = mask_G.sum(axis=0)
mask_C = train_spl == 'C'
count_C = mask_C.sum(axis=0)
gc = 0.423 #GC состав из базы данных NCBI
eps = 0.1
prob_A = (count_A+eps)/(len(train_spl)+eps)
prob_T = (count_T+eps)/(len(train_spl)+eps)
prob_G = (count_G+eps)/(len(train_spl)+eps)
prob_C = (count_C+eps)/(len(train_spl)+eps)
A = np.log(2*prob_A/(1-gc))
T = np.log(2*prob_T/(1-gc))
G = np.log(2*prob_G/gc)
C = np.log(2*prob_C/gc)
pwm = pd.DataFrame([A, T, G, C], ['A', 'T', 'G', 'C'], [i for i in range(1, 14)])
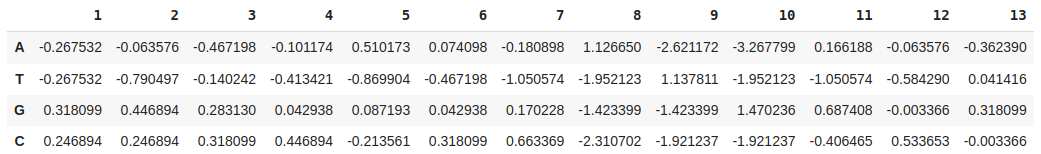
positive = []
for i in test_spl:
positive.append(np.sum([pwm.loc[j].iloc[k] for k, j in enumerate(i)]))
import random
from Bio import SeqIO
neg_df = pd.read_csv('./sarsatg.tsv', sep = '\t')
random.seed(422)
clean_negative = neg_df[(neg_df.Strand == '+') & (neg_df.Pattern != 'start-tr')].reset_index(drop = True)
negative_random = random.choices(clean_negative.index, k = 100)
coords_atg = clean_negative.iloc[negative_random, [0, 1]].reset_index(drop = True)
coords_atg.Start = coords_atg.Start - 8
coords_atg.End = coords_atg.End + 3
iterator = SeqIO.parse('./sars.fasta', 'fasta')
genome = next(iterator)
negative_seqs = []
for i in coords_atg.index:
negative_seqs.append(genome.seq[int(coords_atg.iloc[i, 0]):int(coords_atg.iloc[i, 1])])
with open('./negative_seqs.fasta', 'w') as negative_seqs_fasta:
for i in negative_seqs:
print(i, file = negative_seqs_fasta)
negative = []
for i in negative_seqs:
negative.append(sum([pwm.loc[j].iloc[k] for k, j in enumerate(i)]))
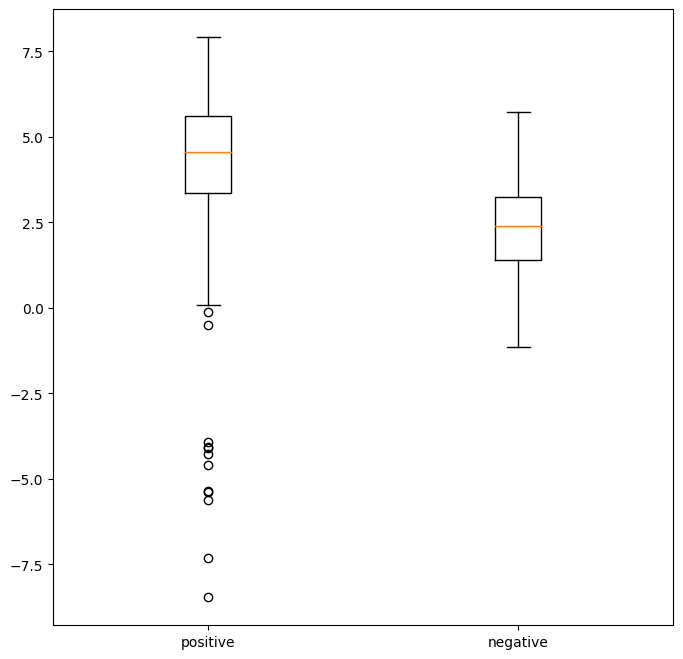
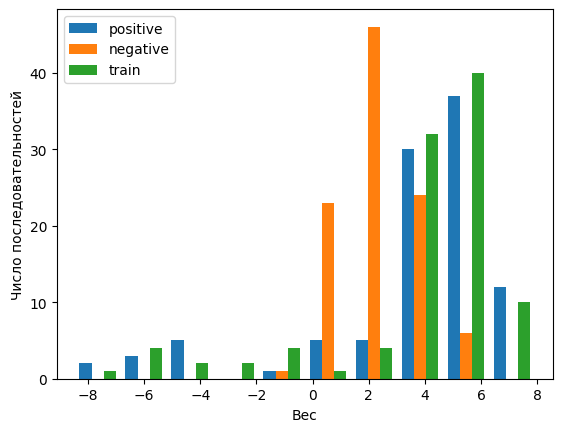
| Обучение | Положительный контроль | Отрицательный контроль | |
|---|---|---|---|
| Сигнал + | 82 | 79 | 30 |
| Сигнал + | 18 | 21 | 70 |
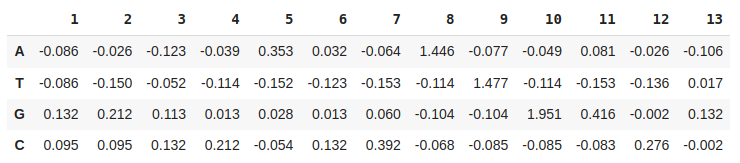
ic_dict = {'A': [], 'T': [], 'G': [], 'C': []}
proba = {'A': (1-gc)/2, 'T': (1-gc)/2, 'G': gc/2, 'C': gc/2}
transposed = np.transpose(list(map(lambda x: [i for i in x.__str__()], train)))
df = pd.DataFrame(transposed)
for i in df.index:
values = df.iloc[i, :].value_counts()
for k, v in proba.items():
try:
ic_dict[k].append(round((values.loc[k]/len(train_spl))*np.log2(values.loc[k]/(len(train_spl)*v)), 3))
except KeyError:
ic_dict[k].append(0)
ic = np.transpose(pd.DataFrame(ic_dict, [i for i in range(1, 14)]))
Литература
1. McLane, L.M. and Corbett, A.H. (2009), Nuclear localization signals and human disease. IUBMB Life, 61: 697-706. https://doi.org/10.1002/iub.194
Благодарности
При выполнение работы мне сильно помогли скрипты Филиппа Качкина, спасибо ему за понятные примеры кода.