AlphaFold2
Предсказание агрегатов амилоидов
Для предсказания стуктуры я взяла последовательность A10:
LLAEPQIAMFCGRLNMHMNVQNGKWDSDPSGTKTCIDTKEGILQYCQEVYPELQITNVVEANQPVTIQNWCKRGRKQCKTHPHFVIPYRCLVGEFV
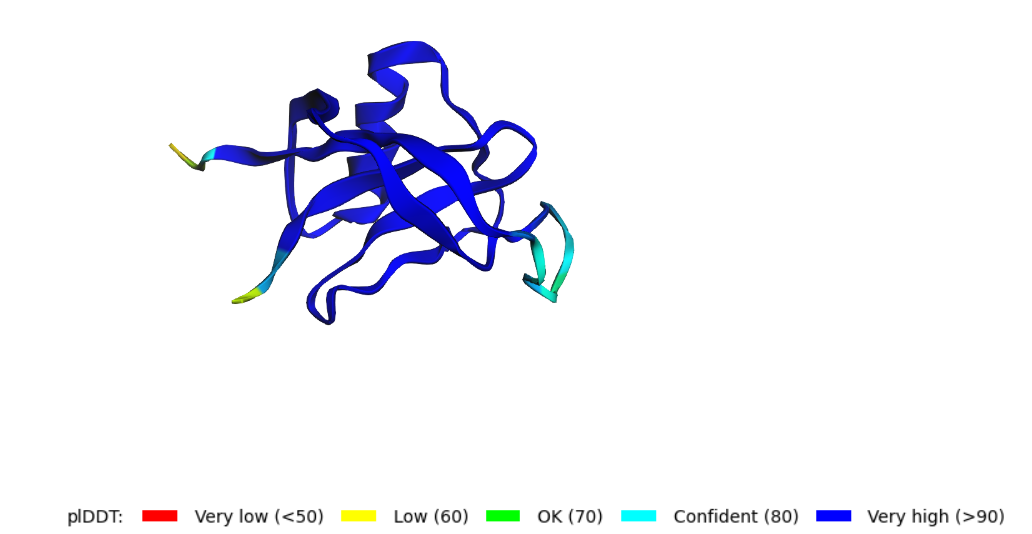
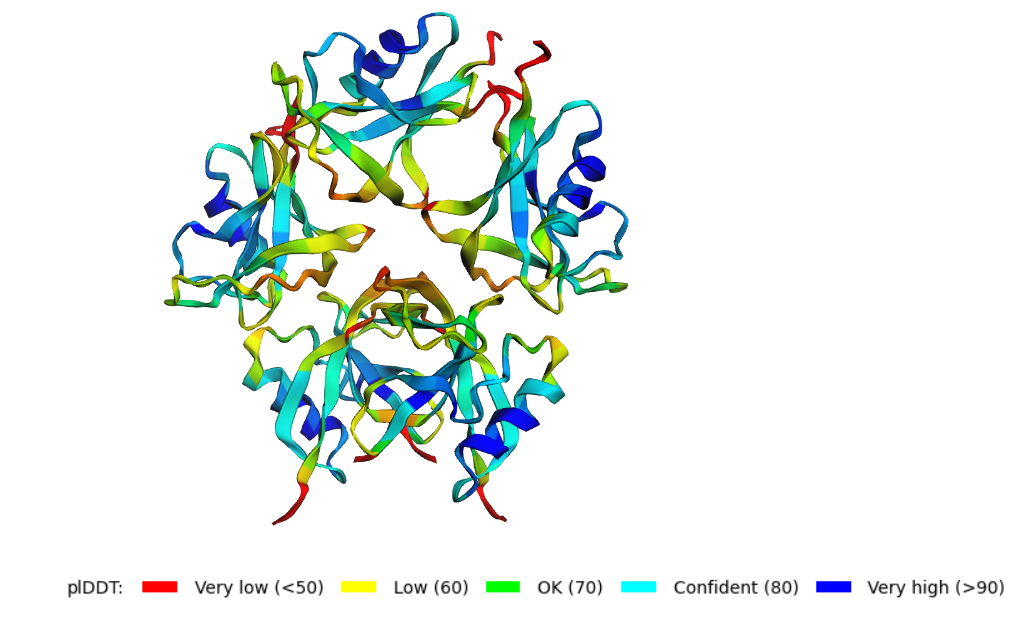
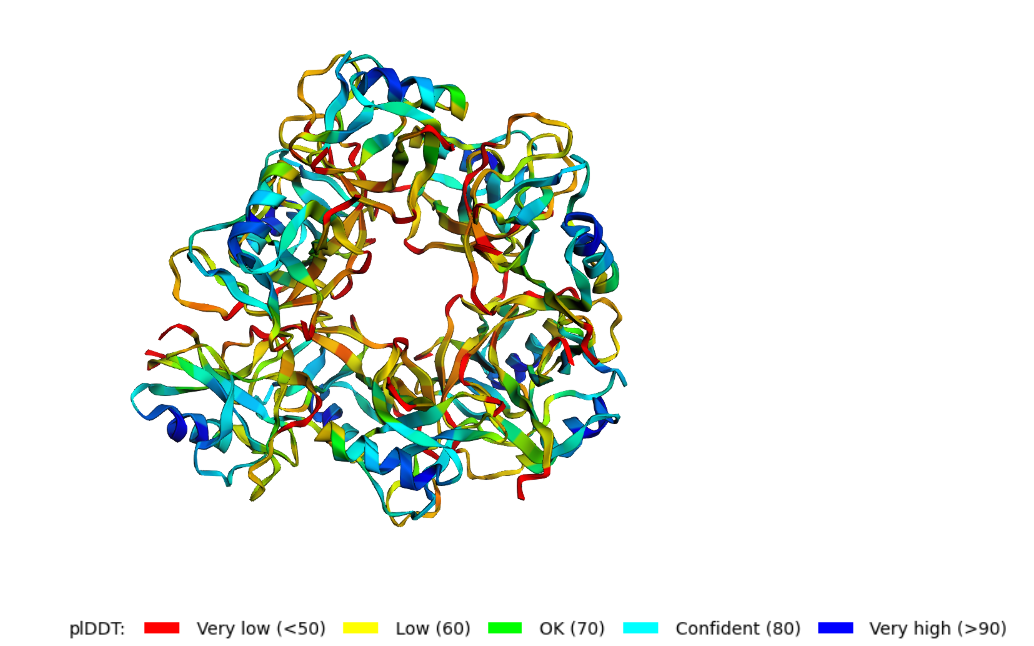
Я сделала предсказания для мономера, пентамера и декамера. Их структуры можно видеть на картинке 1-3.
Рис. 1-3. Предсказание структуры мономера, пентамера и декамера при помощи AlphaFold2.
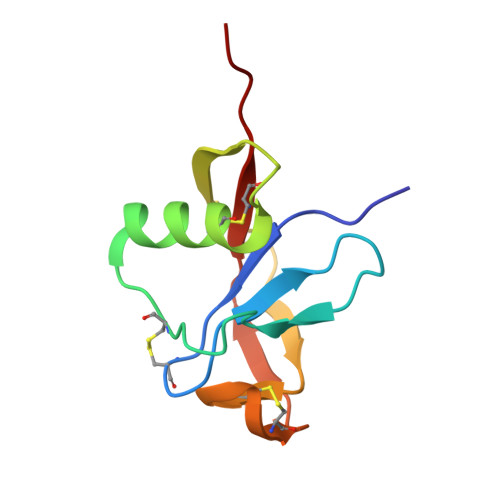
Оказалось, что в pdb этот белок с pdb id 1MWP:
Рис. 4. Структура 1MWP, полученная экспериментально.
AlphaFold2 довольно точно предсказал структуру мономера (подозреваю потому что он на ней учился). Как на счет
пентамера и декамера? В обоих случаях AlphaFold2 попытался склеить бета-листы вместе с образованием "бублика".
Фолдинга в амилоидную фибриллу не наблюдается, то есть предсказазать аггрегацию амилоидных белков AlphaFold2 не удалось.
Алгоритм DOMAK
Для реализации этого алгоритма вначале были посчитаны контакты между остатками аминокислот при помощи arpeggio.
Мне достался белок 1CG2, цепь B. После того как были посчитаны контакты, были отобраны только белок-белковые взаимодействия
внутри этой цепи, после чего для каждого остатка структуры был посчитано split_value*, показывающий насколько
остаток служит "доменным разделителем":
def split_value(split_pos, df):
intA = len(df[(df["bgn.auth_seq_id"]<=split_pos) & (df["end.auth_seq_id"]<=split_pos)])
intB = len(df[(df["bgn.auth_seq_id"]>split_pos) & (df["end.auth_seq_id"]>split_pos)])
extAB = len(df[(df["bgn.auth_seq_id"]<split_pos) & (df["end.auth_seq_id"]>split_pos)])
extAB += len(df[(df["bgn.auth_seq_id"]>split_pos) & (df["end.auth_seq_id"]<split_pos)])
try:
SplitValue=(intA/extAB)*(intB/extAB)
return SplitValue
except ZeroDivisionError:
return 0
*Нумерация всех цепей начиналась не с 1, а с 26, поэтому присутствует try-except блок.
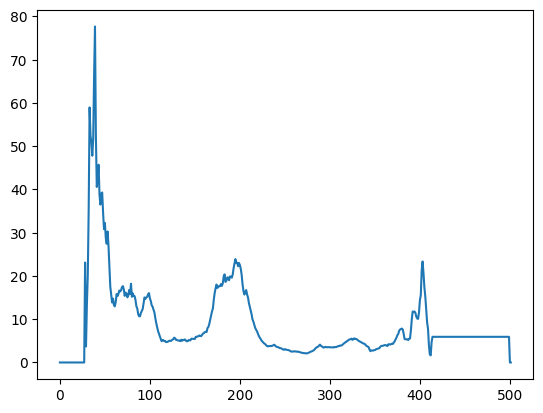
Далее был построен график значений split_value:
Рис. 5. График значений split_value в зависимости от позиции в белке. Первые 23 позиций отсутствуют в структуре, поэтому там нулевое значение.
А теперь посмотрим на соответствие пиков split_value и реальной доменной организации белка. Поскольку пика
3, то предполагаемых домена 4, поскольку пик отвечает аминокислоте, которая располагается на стыке двух доменов.
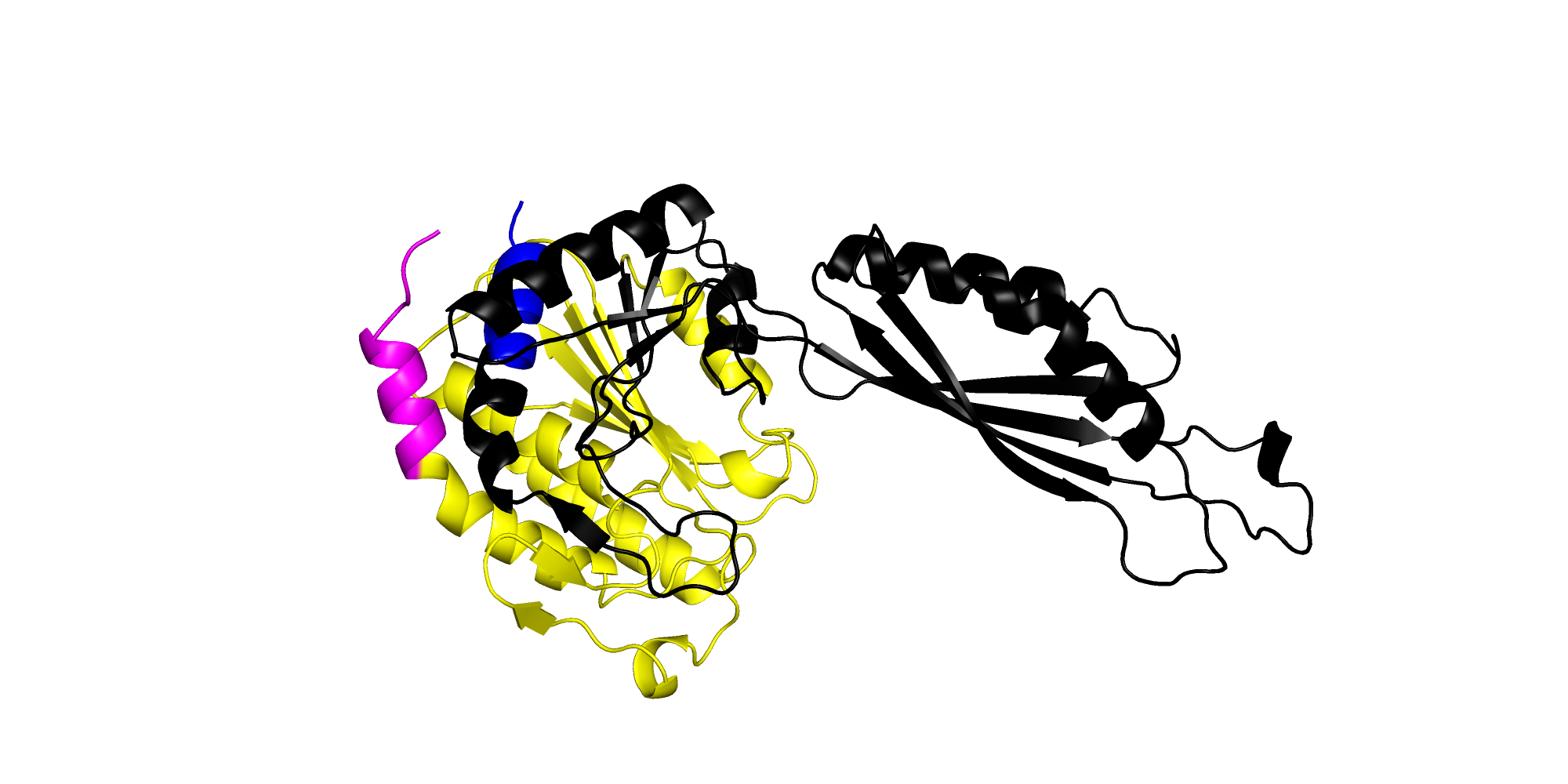
На рис. 6 я выделила эти "домены" разными цветами.
Рис. 6. Структура 1CG2 (цепь B), выделены домены с границами: 23-39 (розовый), 39-195 (желтый), 195-404 (черный).
Видно, что это крайне неудачное предсказание остатков разъединяющих домены.
Из рисунка 6 понятно, что алгоритм не справился с задачей: отчетливо видны минимум два домена соответстувющие архитектурам
α/β (побольше слева и поменьше справа), причем алгоритм запихивает их в один. Кроме того крайние "домены" — терминальные
альфа-спирали. Они обособились видимо по причине того, что внутри альфа-спирали действительно много взаимодействий (как и в остальной
"туловищной части" в целом). Взаимоконтактов "туловища" и "хвоста" также мало по причине терминального положения
спирали. В результате терминальные альфа-спирали по всей видимости сильно портят предсказание такого алгоритма.
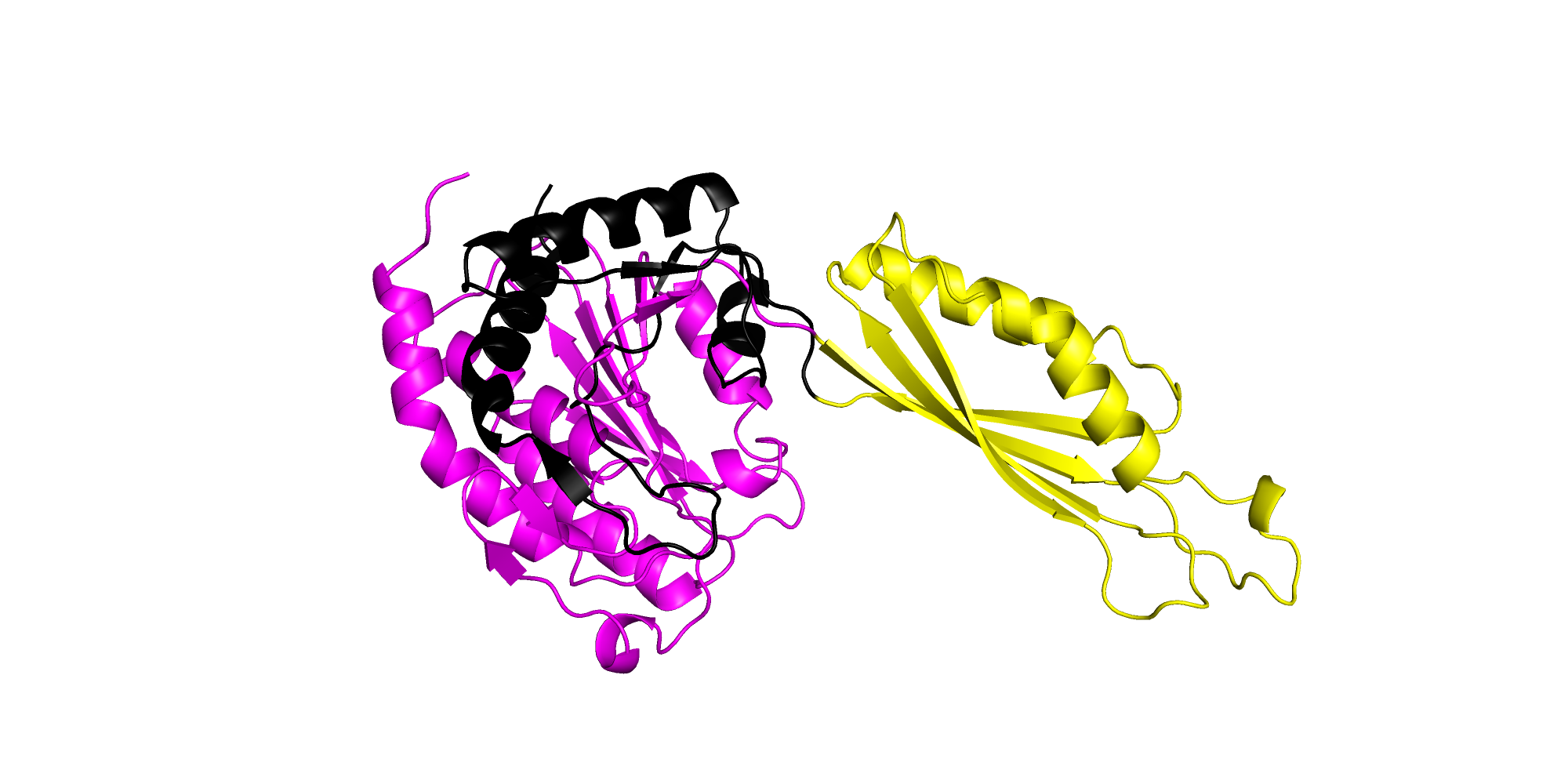
Посмотрим, что говорят базы данных CATH и SCOPE.
Данные базы данных одинаково делят белок на домены (рис. 7). Всего выделяется 3 отдельных домена.
Рис. 7. Структура 1CG2 (цепь B),выделены домены с границами: 23-215 (розовый), 215-324 (желтый), 324-415 (черный).
Я была не права, когда на первый взгляд выделила лишь 2 домена. Оказывается большой "домен" (кавычки, тк это не домен
в строгом смысле слова) по своей сути делится на два структурных домена α/β.