Ресеквенирование. Поиск полиморфизмов у человека.
I часть. Подготовка чтений.
Перед началом работы мною в директорию /nfs/srv/databases/ngs/yudna были скопированы фыйлы:
референсный файл chr4.fasta (сборка версии hg19) и chr4.fastq.
1. Проведение анализа качества чтения. Необходимо сделать контроль качества чтения с помощью
программы fastqc. Я использовала программу, установленную на kodomo.
Команда: fastqc chr4.fastq
Выдача: отчет о программе в виде html файла chr4_fastq.html.
2. Очистка чтения. Очистка проводилась с помощью программы trimmomatic, установленной на
kodomo. Требовалось отрезать с конца каждого прочтения нуклеотиды с качеством ниже 20 (TRAILING:20)
и удалисть прочтения с длинной меньше 50 (MINLEN:50).
Команда: java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr4.fastq chr4_1.fastq
TRAILING:20 MINLEN:50
После повторного анализа качества прочтения был получен файл chr4_1_fastqc(2).html .
Сравнение.
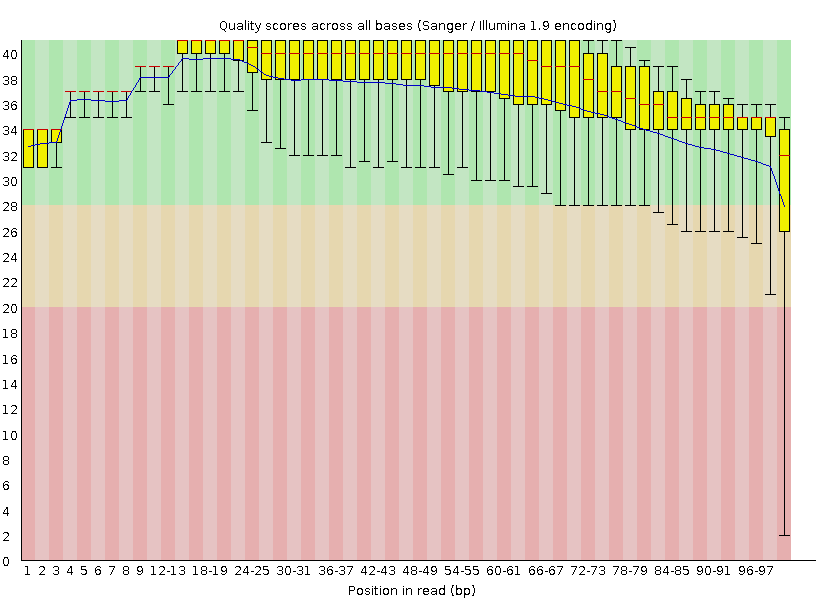
Рис.1. 'Per base quality' до чистки
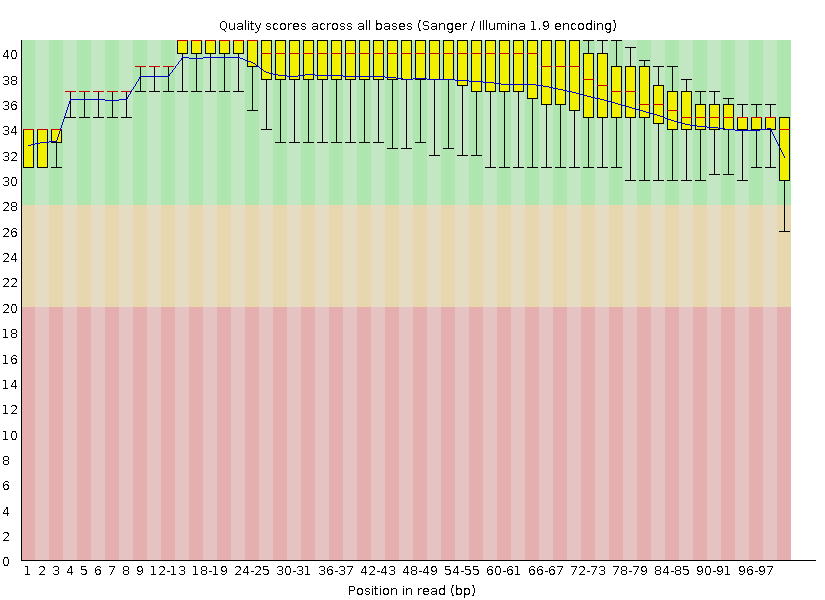
Рис.2. 'Per base quality' после чистки
На рис.1 и 2 представлены графики качества чтения, полученные в программе fastqc. Синяя линия соответсвует
среднему качеству чтения, красная линия - медиане, желтые столбики - интеркалярному размаху (разнице между верхним и нижним квартилями).
Поле графика поделено на три области - зеленую, желктую и красную, попадание в которые разных элементов графика
позволяет судить о качестве их прочтения.
По графикам видно, что качетво прочтения улучшилось после работы программы trimmomatic -
практически все элементы содержаться в зеленой области. В результате чистки было удалено 95 прочтений с длиной меньше 50, общая сумма теперь
составляет 5715 с длиной от 50 до 100. До чистки было 5810 прочтений с длиной 43-100.
II часть. Картирование чтений.
3. Картирование чтений. Данный процесс подразделяется на наесколько этапов. Сначала надо
индексировать референсную последовательность, затем построить выравнивания прочтений и референсной полседовательности.
Таблица 1
| Команда |
Назначение/Выдача |
| export PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5 |
Вызывает программу, лежащую в указанной директории. |
| hisat2-build chr4.fasta chr4 |
Индексирует референснуб последовательность, выдает множество файло с расширением .ht2 |
| hisat2 -x chr4 -U chr4_1.fastq --no-spliced-alignment --no-softclip > align_1.sam |
Строит выранивание прочтения и референса, сохраняет результаты в отдельный файл align_1.sam |
4. Анализ выравнивания. Для анализа выравнивания использовалась программа sаmtools. Результаты ее работы
сведены в таблицу.
Таблица 2
| Команда |
Назначение/Выдача |
| samtools view align_1.sam -bo align_1.bam |
Переводит выравние с референсом в бинарный формат |
| samtools sort align_1.bam -T myfile.txt -o sorted_aln.bam |
Сортирует выравнивание чтений с референсом по координате в референсе начала чтения. |
| samtools index sorted_aln.bam |
Индексирует отсортированный файл .bam |
Количесво откартированных на референсную последовательность чтений можно выяснить двумя способами:
- После работы программы Hisat2, на stdout выдыется информация о прочтениях: 19 прочтений не были выравнены совсем, 5694 - были выравнены 1 раз, 2 - были выравнены более 1 раза.
- Работа с файлом .bam. Команда: samtools idxstats sorted_aln.bam > stats.txt. Выдача stats.txt.
Таким образом может быть получена информация лишь о количестве откартированных прочтений.
III часть. Анализ SNP.
5. Поиск SNP и инделей. Для этих целей вновь использовалась программа samtools и пакет программ bcftools.
Таблица 3
| Команда |
Назначение/Выдача |
| samtools mpileup -uf chr4.fasta sorted_aln.bam > snp.bcf |
Создает файл с полиморфизмами на основе референсной последовательности и файла с выравниванием прочтений. |
| bcftools call -cv snp.bcf > snp.vcf |
Создает список отличий между референсом и чтениями. Файл: snp.vcf. |
По результатам выдачи программы bcftools, было найдено 49 полиморфизмов, из которых 4 инделя и 45 замен. Ниже описаны три полиморфизма.
В таблице найденные полиморфизмы ранжированы по качеству прочтени я и качеству покрытия, видно, что у первой вставки эти параметры самые хорошие.
Таблица 4
| Координата |
Тип полиморфизма |
Референс |
Прочтение |
Глубина |
Качество |
| 68458937 |
Замена |
G |
C |
87 |
225.009 |
| 88760642 |
Вставка |
AAGAGA |
AAGAGAGA |
16 |
81.4666 |
| 187165891 |
Делеция |
GTTTTT |
GTTTT |
2 |
3.66479 |
6. Аннотация SNP. С помощью программы annovar и баз данных refgene, dbsnp, 1000 genomes, GWAS, Clinvar
требуется проаннотировать snp, предварительно исключив все индели.
Команда: perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 /nfs/srv/databases/ngs/yudna/snp.vcf >
/nfs/srv/databases/ngs/yudna/snp.avinput
Выдача: Файл, готовый для использования annovar: snp.avinput.
Поиск по базам данных:
-
- Аннотация по dbsnp
- Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.snp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb/
- Из 45 полиморфизмов 39 имеют rs и 6 не имеют. rs.snp.hg19_snp138_dropped, rs.snp.hg19_snp138_filtered.
-
- Аннотация по refgene
- Команда:perl /nfs/srv/databases/annovar/annotate_variation.pl -out rs.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb/
- Получены два файла, содержащие информацию по полиморфизмам - rs.refgen.varient_function
и rs.refgen.exonic_varient_function.
В файле .varient_function snp разделены по группам в зависимости от положения в геноме (Таблица 5). SNP также
разделяются на het и hom замены: 16 het и 33 hom.
Таблица 5
| intronic |
exonic |
introgenic |
UTR3 |
downstream |
| 40 |
3 |
3 |
1 |
2 |
Из таблицы видно, что наибольшее количество замен приходится на интроны. Это можно объяснить тем, что изменения в этих
последовательностях не подвержены действию отбора, так как не влияют на конечный продукт гена. В таблицу 6 собрана информация об изменениях в экзонах.
Таблица 6
| Координата |
Ген (экзон) |
Тип замены |
Качество прочтения |
Глубина прочтения |
Было -> Стало |
| 187158034 |
KLKB1 exon5 |
Несинонимичная |
221.999 |
83 |
G -> A |
| 187172943 |
KLKB1 exon10 |
Несинонимичная |
225.009 |
66 |
A -> G |
| 187179210 |
KLKB1 exon15 |
Синонимичная |
225.009 |
31 |
T -> C |
Данный ген KLKB1 кодирует гликопротеин, который участвует в процессе свертывания крови.
-
- Аннотация по Clinvar
- Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.clinvar -dbtype clinvar_20150629 -buildver hg19 snp.avinput /nfs/srv/databases/annovar/humandb/
- Получены два файла: _dropped и _filtered. В первом содержится информация о ранее описанных полиморфизмах -
замена нуклеотида в экзоне 5 KLKB1 приводит к дифециту прекалликреина (плазменного компонента крови). Остальные полиморфизмы содержатся во втором файле и не аннотированы.
-
- Аннотация по 1000Genomes
- Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.1000g -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb/
- Поиск по этой БД дает информацио о частоте встречаемости анализируемых полиморфизмов. Самая высокая - 0.91853;
самая низкая - 0.00379393. Частота встречаемости замены, описанной в клинической БД - 0.604633.
-
- Аннотация по GWAS (Genome-Wide Association Studies)
- Команда: perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out rs.gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb/
- Поиск по этой БД дает информацию о связи полиморфизмов с фенотипическими проявлениями. Однако для встетившихся полиморфизмов файл, выданный программой, оказался пустым,
то есть нет ни одного описанного фенотипического проявления.
Источники
FastQC
Trimmomatic
Hisat2
Annovar
|